Atypical Teratoid/Rhabdoid Tumour (AT/RT)¶
Summary
- Rare, highly aggressive embryonal tumour of the central nervous system (CNS)
- Primarily affects young children, typically under 3 years of age
- Characterised by loss of INI1/SMARCB1 gene expression and distinctive imaging features
Pathophysiology¶
- Arises from embryonal cells in the CNS
- Defined by biallelic inactivation of SMARCB1 gene (95% of cases) or SMARCA4 gene (2% of cases)
- Loss of these genes leads to dysregulation of chromatin remodelling and cell cycle control
- Histologically heterogeneous, containing rhabdoid cells, primitive neuroectodermal cells, and mesenchymal elements
Demographics¶
- Accounts for 1-2% of paediatric brain tumours
- Median age at diagnosis: 17 months
- Slight male predominance (1.6:1)
- Rare cases reported in adults
Diagnosis¶
- Clinical presentation:
- Rapid onset of neurological symptoms
- Increased intracranial pressure
- Focal neurological deficits
- Laboratory findings:
- CSF cytology may show malignant cells
- Histopathology:
- Characteristic loss of INI1/SMARCB1 protein expression on immunohistochemistry
- Presence of rhabdoid cells with eccentric nuclei and eosinophilic cytoplasmic inclusions
Imaging¶
- CT:
- Hyperdense, heterogeneous mass
- Often with areas of haemorrhage and necrosis
- Calcifications in 40-60% of cases
- MRI:
- T1: Heterogeneous, predominantly iso- to hypointense
- T2: Heterogeneous, predominantly hyperintense
- T1 post-contrast: Heterogeneous enhancement
- DWI: Restricted diffusion in solid components
- MR spectroscopy: Elevated choline, reduced NAA, presence of lipid/lactate peaks
- Location:
- 50-60% infratentorial (cerebellum, brainstem)
- 40-50% supratentorial
- Rarely in the spinal cord
- Distinctive features:
- "Cyst and nodule" appearance in some cases
- Frequent leptomeningeal dissemination at diagnosis (20-30%)
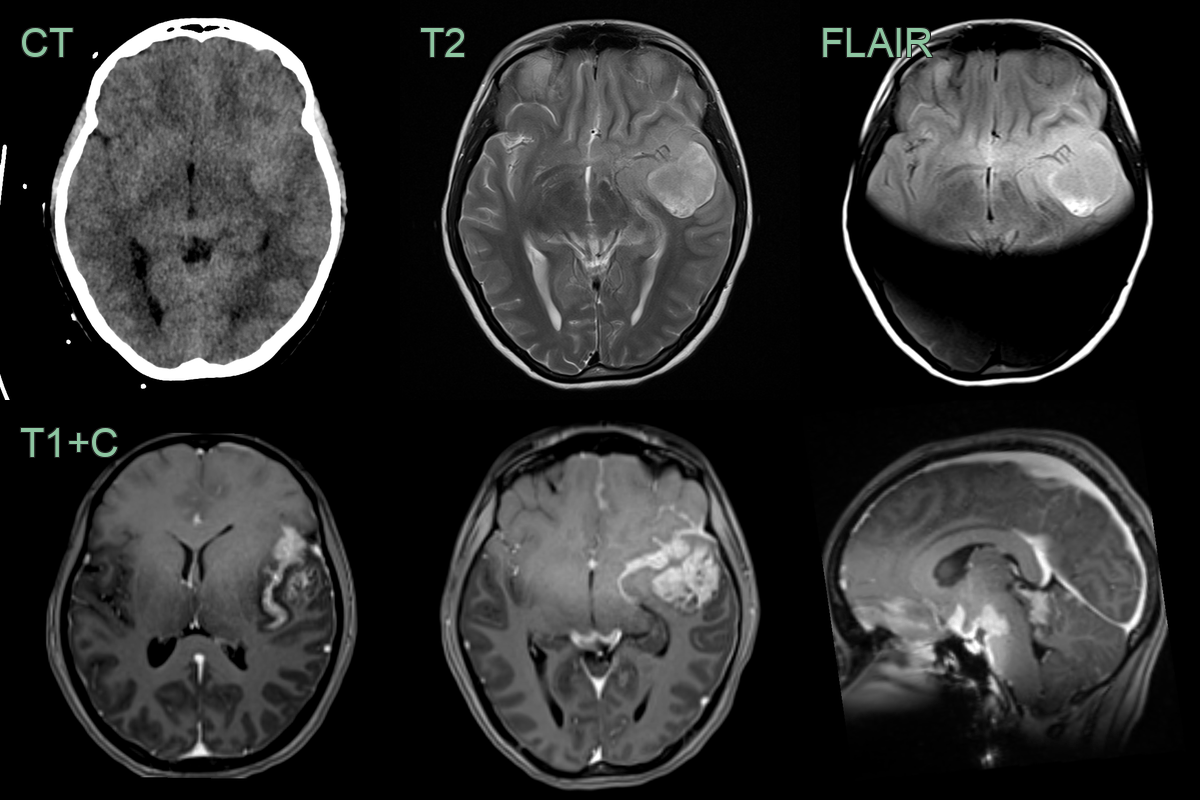
- A 15-year-old patient presenting with seizures and headache.
- CT showed only subtle hyperdensity in the left anterior temporal lobe and surrouding sulci.
- MRI showed an heterogeneously enhancing tumour in the left anterior frontal lobe with extensive leptomeningeal disease in the anterior, middle and posterior cranial fossae.
Treatment¶
- Multimodal approach:
- Maximal safe surgical resection
- Intensive chemotherapy
- Craniospinal irradiation (in children >3 years)
- Novel therapies under investigation:
- EZH2 inhibitors
- CDK4/6 inhibitors
- Immunotherapy approaches
- Prognosis:
- Poor, with median survival of 6-12 months
- 5-year overall survival: 15-30%
- Factors associated with better prognosis: older age at diagnosis, gross total resection, and supratentorial location
Differential diagnosis¶
| Differential Diagnosis | Distinguishing Feature |
|---|---|
| Medulloblastoma | ATRT typically has a more heterogeneous appearance on MRI and often involves the cerebellopontine angle |
| Choroid plexus carcinoma | ATRT tends to have a more aggressive clinical course and often presents in younger patients |
| Primitive neuroectodermal tumour (PNET) | ATRT shows loss of INI1/SMARCB1 expression on immunohistochemistry |
| Ependymoma | ATRT typically has a more heterogeneous enhancement pattern and often lacks the "plastic" appearance of ependymomas |
| Glioblastoma | ATRT more commonly occurs in infants and young children, while glioblastoma is more common in older children and adults |
| Teratoid tumour | ATRT has a characteristic loss of INI1/SMARCB1 expression, which is not seen in teratoid tumours |
| Metastatic neuroblastoma | ATRT is typically a primary CNS tumour, while neuroblastoma metastases are secondary |
| Embryonal tumour with multilayered rosettes (ETMR) | ATRT lacks the characteristic C19MC amplification seen in ETMR |