Chordoma¶
Summary
- Rare, slow-growing malignant tumour arising from notochordal remnants
- Typically occurs in the axial skeleton, most commonly at the sacrum and skull base
- Characterised by locally aggressive behaviour and high recurrence rates
Pathophysiology¶
- Originates from persistent notochordal remnants along the axial skeleton
- Expresses brachyury, a key transcription factor in notochord development
- Three histological subtypes:
- Conventional (most common)
- Chondroid
- Dedifferentiated (most aggressive)
Demographics¶
- Incidence: 0.08 per 100,000 person-years
- Median age at diagnosis: 58-60 years
- Slight male predominance (male-to-female ratio 1.5:1)
- Distribution by location:
- Sacrococcygeal: 50-60%
- Skull base: 25-35%
- Mobile spine: 15%
Diagnosis¶
- Clinical presentation:
- Sacral: pain, neurological deficits, bowel/bladder dysfunction
- Skull base: cranial nerve palsies, headache, visual disturbances
- Histopathology:
- Physaliphorous cells with vacuolated cytoplasm
- Positive immunohistochemistry for brachyury, cytokeratin, and S100 protein
- Genetic testing:
- Duplication of brachyury gene (T) on chromosome 6q27
Imaging¶
- CT:
- Lytic, destructive lesion with soft tissue mass
- Calcifications in 30-70% of cases
- MRI:
- T1: hypointense to isointense
- T2: hyperintense with heterogeneous signal
- Strong enhancement with gadolinium
- "Honeycomb" appearance due to fibrous septations
- PET/CT:
- Variable FDG uptake, more useful for metastatic disease detection
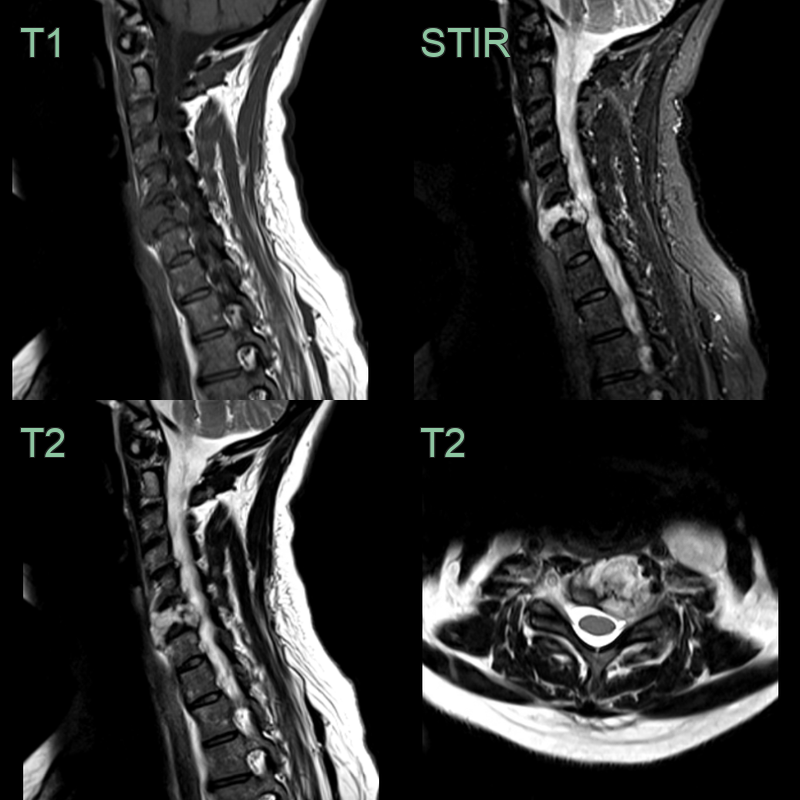
- 50-year-old patient presented with neck pain and a left arm radiculopathy.
- A hyperintense and expansile lesion filling the C6 vertebral body caused compression of the left C7 nerve root.
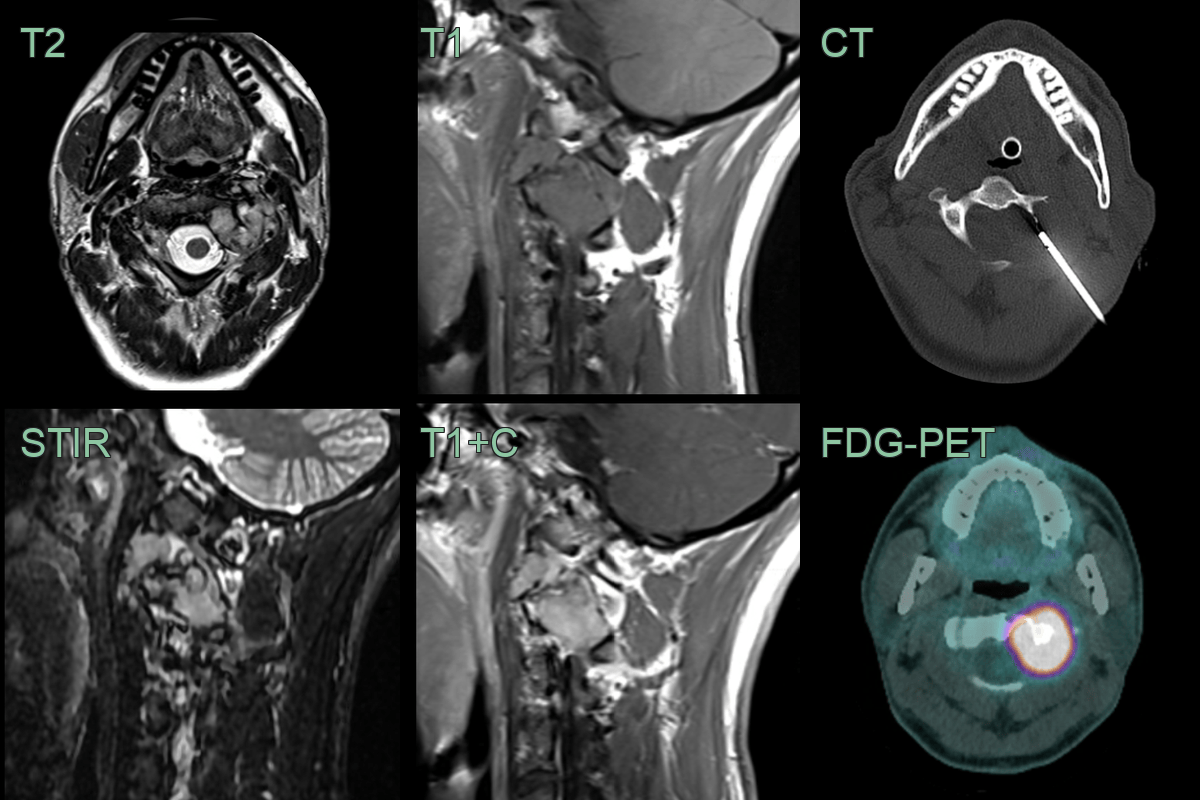
- 30-year-old patient 3 month history of neck pain and crepitus.
- MRI showed an expansile enhancing lesion centred on the left side of the C3 vertebra.
- The lesion was lucent on CT and metabolically active on FDG-PET.
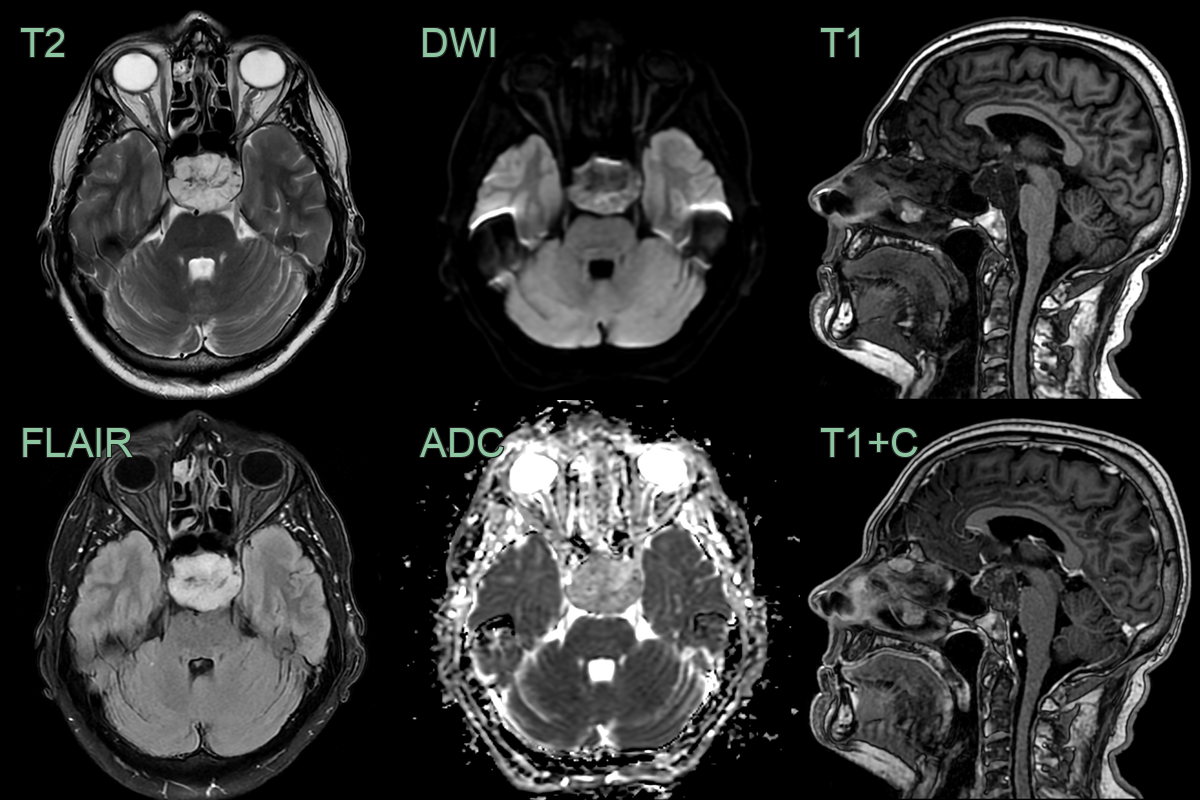
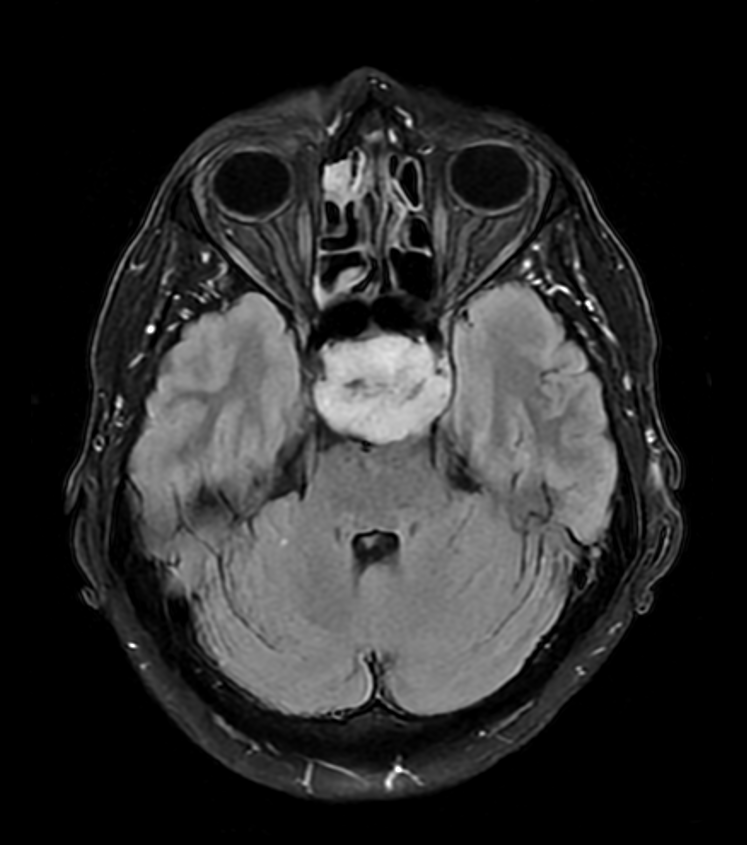
- 60-year-old patient presented with visual impairment and clinical features of hypopituitarism.
- MRI showed a minimally enhancing lesion replacing the superior clivus and pititary fossa.
- The pituitary gland, infundibular stalk and optic chiasm were compressed.
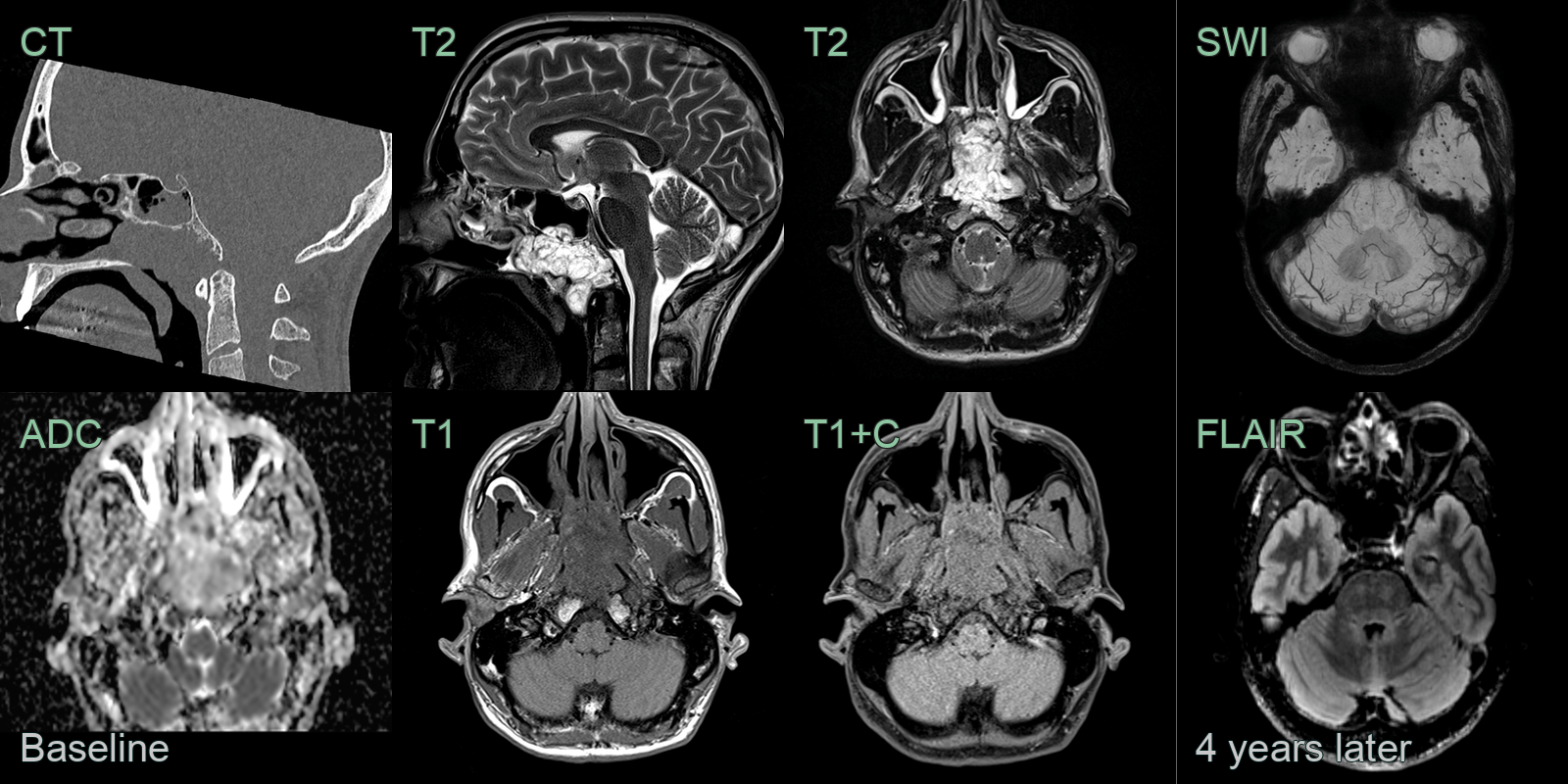
- A 50-year-old patient presented with nasal obstruction.
- MRI showed a lobulated mild enhancing lesion in the nasopharynx with erosion of the inferior cortex of the clivus.
- 4 years later, a follow-up MRI showed no recurrence but many microhaemorrhages in the anterior temporal lobes and brainstem, which were likely to be related to radiotherapy.
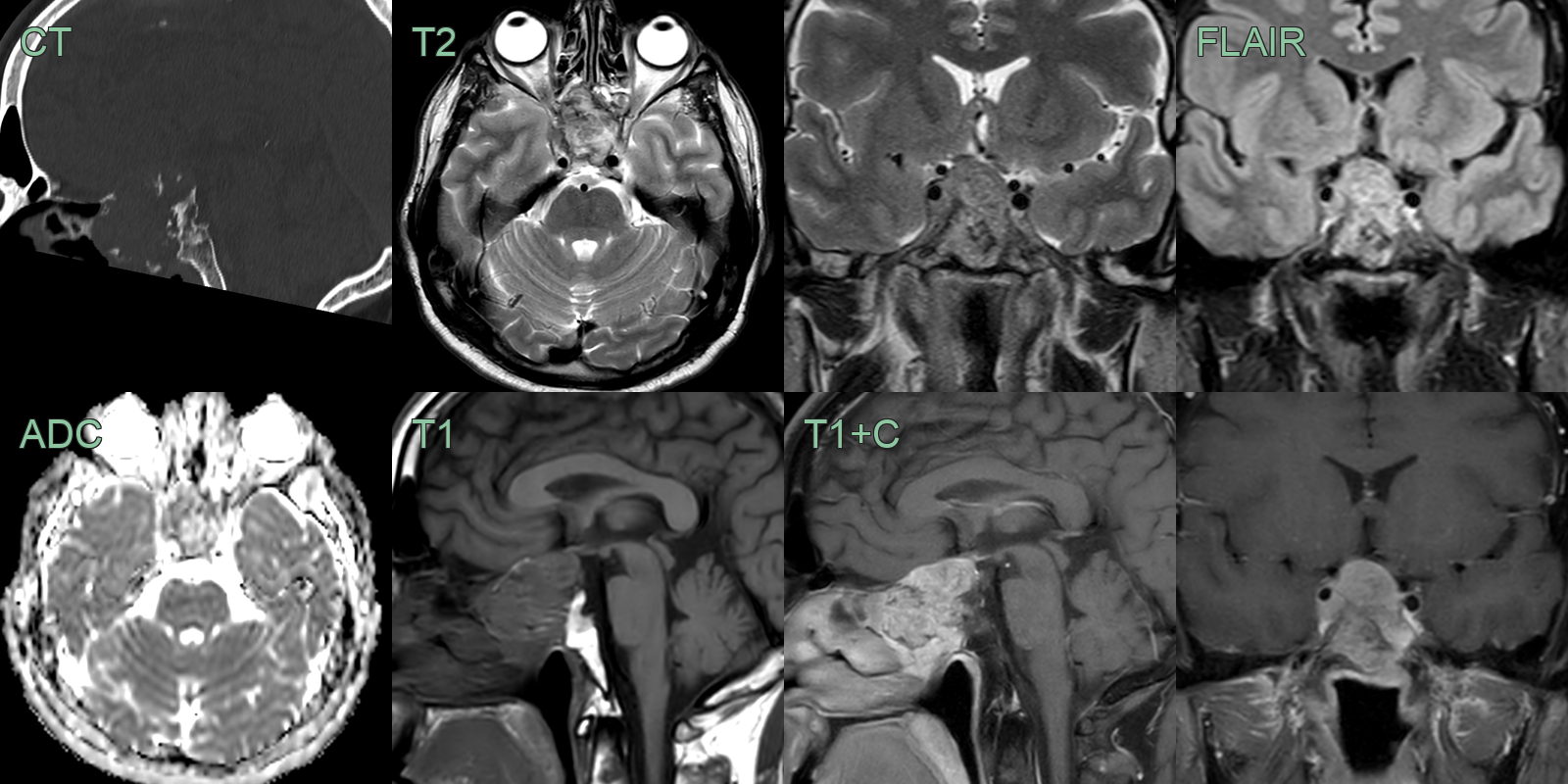
- A 50-year-old patient presented with a visual field defect picked up during a routine eye test.
- CT showed a large destructive lesion centred on the anterior clivus and pituitary fossa.
- MRI showed an avidely enhancing clivus lesion that was compressing the right cisternal optic nerve.
- With the differential including a pituitary macroadenoma, a chordoma was confirmed following a transphenoidal biopsy.
Treatment¶
- Surgery:
- En bloc resection with wide margins is the primary treatment
- Challenging due to proximity to critical structures
- Radiation therapy:
- Adjuvant or definitive treatment
- Proton beam therapy or carbon ion therapy for improved local control
- Systemic therapy:
- Limited efficacy of conventional chemotherapy
- Targeted therapies:
- Imatinib for PDGFR-positive tumours
- Erlotinib for EGFR-positive tumours
- Immunotherapy:
- Ongoing clinical trials with checkpoint inhibitors
Differential diagnosis¶
| Differential Diagnosis | Differentiating Feature |
|---|---|
| Chondrosarcoma | Lacks the physaliphorous cells characteristic of chordoma; typically shows chondroid matrix |
| Metastatic carcinoma | Usually lacks the myxoid stroma seen in chordoma; immunohistochemistry differs |
| Pituitary adenoma | Typically confined to the sella turcica; lacks notochordal differentiation |
| Meningioma | Usually dural-based; lacks physaliphorous cells; positive for EMA and PR |
| Schwannoma | Typically encapsulated; S100 positive but brachyury negative |
| Ecchordosis physaliphora | Small, incidental finding; lacks invasive growth pattern of chordoma |
| Giant cell tumour | Lacks physaliphorous cells; contains numerous osteoclast-like giant cells |
| Osteosarcoma | Produces osteoid matrix; lacks physaliphorous cells and myxoid stroma |
| Paraganglioma | Shows characteristic "zellballen" pattern; positive for neuroendocrine markers |
| Ependymoma | Typically intraventricular; shows perivascular pseudorosettes; GFAP positive |