Fabry Disease¶
Summary
- X-linked lysosomal storage disorder caused by deficiency of α-galactosidase A enzyme
- Progressive accumulation of glycosphingolipids in various tissues and organs
- Characterised by neuropathic pain, angiokeratomas, renal and cardiac dysfunction
Pathophysiology¶
- Mutation in GLA gene on X chromosome (Xq22)
- Deficiency of α-galactosidase A enzyme leads to:
- Accumulation of globotriaosylceramide (Gb3) in lysosomes
- Progressive damage to vascular endothelium, kidneys, heart, and nervous system
- Multisystemic involvement due to widespread glycosphingolipid deposition
Demographics¶
- Incidence: 1:40,000 to 1:117,000 live births
- X-linked inheritance pattern:
- Males more severely affected
- Females can be carriers or have variable disease expression
- Onset:
- Classic form: childhood or adolescence
- Late-onset variants: adulthood
Diagnosis¶
- Clinical presentation:
- Acroparesthesias and neuropathic pain
- Angiokeratomas
- Corneal opacities (cornea verticillata)
- Hypohidrosis or anhidrosis
- Proteinuria and progressive renal failure
- Left ventricular hypertrophy and arrhythmias
- Laboratory tests:
- Decreased α-galactosidase A enzyme activity in plasma or leukocytes
- Elevated plasma and urinary Gb3 levels
- Genetic testing:
- GLA gene sequencing to confirm diagnosis and identify specific mutation
Imaging¶
- Cerebral MRI:
- White matter lesions
- Dolichoectasia of basilar artery
- Increased signal intensity in pulvinar on T1-weighted images ("pulvinar sign")
- Cardiac imaging:
- Echocardiography: Left ventricular hypertrophy, valvular abnormalities
- Cardiac MRI: Late gadolinium enhancement in basal inferolateral wall
- Renal imaging:
- Ultrasound: Increased echogenicity, cysts, and reduced corticomedullary differentiation
- MRI: T1 and T2 shortening due to lipid accumulation
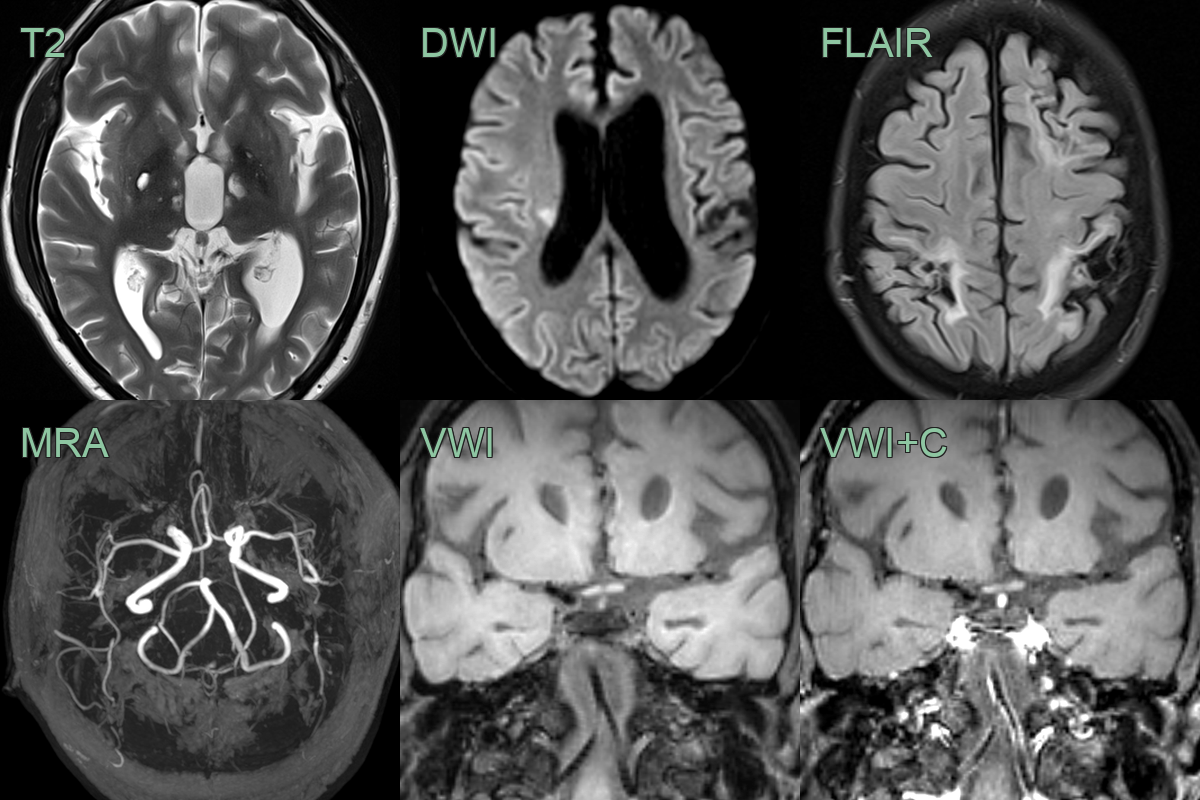
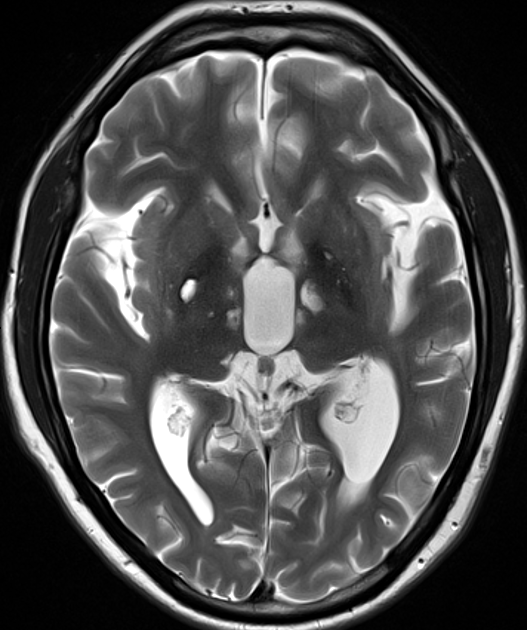
- 30-year-old patient with history of Fabry disease presented with left sided weakness.
- There was an acute infarct in the rigth corona radiata as well as many old small vessel deep lacunar infarcts and larger vessel infarcts in both cerebral hemispheres.
- Vessel wall imaging showed eccentric enhancement of the vertebrobasilar system without dolichoectasia.
Treatment¶
- Enzyme replacement therapy (ERT):
- Agalsidase alfa or agalsidase beta
- Intravenous infusion every two weeks
- Slows disease progression and improves quality of life
- Chaperone therapy:
- Migalastat for amenable GLA mutations
- Supportive care:
- Pain management
- ACE inhibitors or ARBs for proteinuria
- Anticoagulation for stroke prevention
- Cardiac medications for arrhythmias and heart failure
- Renal replacement therapy:
- Dialysis or kidney transplantation for end-stage renal disease
- Genetic counseling for affected individuals and family members
Differential diagnosis¶
| Differential diagnosis | Differentiating feature |
|---|---|
| CADASIL | Similar small vessel disease white matter pattern with anterior temporal lobe predominance; NOTCH3 mutation; no pulvinar T1 hyperintensity or dolichoectasia |
| Multiple sclerosis | Periventricular ovoid lesions (Dawson's fingers); calloso-septal interface; no T1 pulvinar hyperintensity; no vertebrobasilar dolichoectasia |
| Hypertensive microangiopathy | Deep white matter and basal ganglia hyperintensities; associated with hypertension; no pulvinar sign and no dolichoectasia |
| Mitochondrial disease (MELAS) | Basal ganglia and thalamic signal changes; stroke-like lesions not conforming to vascular territories; lactate peak on MR spectroscopy |
| Cerebral amyloid angiopathy | Lobar microhaemorrhages with posterior predominance; cortical superficial siderosis; typically older patients; no pulvinar sign |