Ganglioglioma¶
Summary
- Rare, slow-growing neuroepithelial tumour composed of neoplastic glial and neuronal cells
- Most commonly occurs in children and young adults, typically presenting with seizures
- Characteristic imaging findings include a cystic mass with a mural nodule, often in the temporal lobe
Pathophysiology¶
- Mixed neuronal-glial tumour with WHO grade 1 classification
- Composed of dysplastic ganglion cells and neoplastic glial cells, typically astrocytic
- Molecular alterations:
- BRAF V600E mutation in approximately 50% of cases
- CD34 expression in most cases
- IDH½ mutations are rare
Demographics¶
- Accounts for 0.4-1.3% of all central nervous system (CNS) tumours
- Peak incidence in children and young adults (median age 20-30 years)
- Slight male predominance (male:female ratio 1.2:1)
- Most common location: temporal lobe (70%), followed by frontal and parietal lobes
Diagnosis¶
- Clinical presentation:
- Seizures (most common, 80-90% of cases)
- Headaches
- Focal neurological deficits
- Histopathology:
- Biphasic pattern with neuronal and glial components
- Dysplastic neurons with abnormal clustering and orientation
- Perivascular lymphocytic cuffing
- Immunohistochemistry:
- Neuronal markers: synaptophysin, NeuN
- Glial markers: GFAP
- CD34 positivity in most cases
Imaging¶
- CT findings:
- Hypodense or isodense mass
- Calcifications in 30-50% of cases
- Variable contrast enhancement
- MRI findings:
- T1: hypointense to isointense
- T2/FLAIR: hyperintense
- Cystic component with mural nodule in 50-60% of cases
- Variable gadolinium enhancement, often in the solid component
- Minimal perilesional oedema
- Advanced imaging:
- MR spectroscopy: elevated choline, decreased NAA
- Perfusion imaging: generally low relative cerebral blood volume
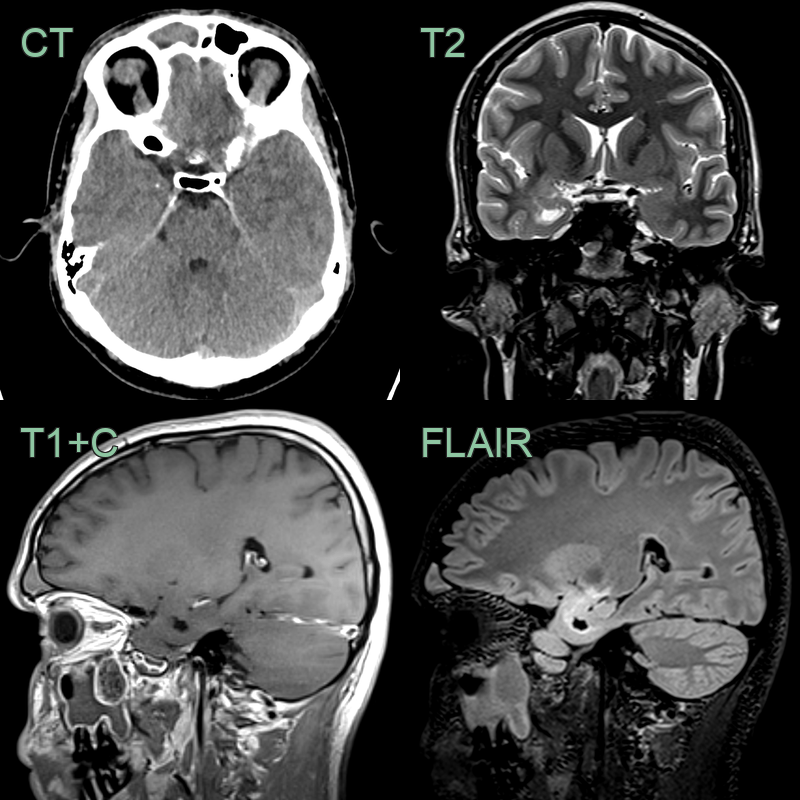
- 20-year-old patient presented with generalised tonic-clonic seizures.
- Imaging showed a non-enhancing solid-cystic lesion in the right mesial temporal lobe with a single speck of calcification.
- Histopathology following resection confirmed a ganglioglioma.
Treatment¶
- Surgical resection is the primary treatment
- Gross total resection associated with better outcomes
- Seizure control achieved in 70-90% of cases after complete resection
- Adjuvant therapy:
- Radiotherapy may be considered for incomplete resection or anaplastic features
- Chemotherapy role is limited, mainly for recurrent or progressive disease
- Targeted therapy:
- BRAF inhibitors (e.g., vemurafenib) show promise in BRAF V600E mutated cases
- Prognosis:
- Generally favourable with 5-year survival rates >90%
- Anaplastic transformation occurs in <5% of cases
- Long-term follow-up is necessary due to potential for late recurrence
Differential diagnosis¶
| Differential Diagnosis | Distinguishing Feature |
|---|---|
| Dysembryoplastic Neuroepithelial Tumour (DNET) | "Bubbly" or multinodular appearance; contrast enhancement uncommon; no mural nodule |
| Pleomorphic Xanthoastrocytoma | Prominent contrast enhancement; dural tail sign; typically superficial location |
| Oligodendroglioma | Calcifications common; "honeycomb" cortical pattern; more diffuse T2 signal |
| Pilocytic Astrocytoma | More common in cerebellum; prominent enhancing mural nodule; Rosenthal fibres on histology |
| Focal Cortical Dysplasia | No mass effect; cortical thickening with blurring of grey-white matter junction; no enhancing nodule |
| Desmoplastic Infantile Ganglioglioma | Large cystic mass with dural involvement; typically in infants under 2 years |