Huntingdon's disease¶
Summary
- Autosomal dominant neurodegenerative disorder characterised by progressive motor, cognitive, and psychiatric symptoms
- Caused by CAG trinucleotide repeat expansion in the huntingtin (HTT) gene
- Imaging shows characteristic striatal atrophy and white matter changes
Pathophysiology¶
- Mutation in HTT gene on chromosome 4p16.3
- CAG repeat expansion leads to production of mutant huntingtin protein
- Accumulation of mutant protein causes neuronal dysfunction and death
- Preferential degeneration of medium spiny neurons in the striatum
- Widespread cortical and subcortical atrophy as disease progresses
Demographics¶
- Prevalence: 5-10 per 100,000 in Western populations
- Age of onset: typically 30-50 years, but can occur at any age
- Juvenile-onset HD (< 20 years) accounts for 5-10% of cases
- No gender predilection
- Higher prevalence in populations of European descent
Diagnosis¶
- Clinical features:
- Motor symptoms: chorea, dystonia, bradykinesia
- Cognitive decline: executive dysfunction, memory impairment
- Psychiatric symptoms: depression, anxiety, irritability
- Genetic testing: CAG repeat expansion ≥ 36 repeats is diagnostic
- Family history: often positive due to autosomal dominant inheritance
- Neurological examination and cognitive assessment
Imaging¶
- MRI findings:
- Striatal atrophy (caudate and putamen)
- Cortical atrophy, particularly in frontal and temporal lobes
- White matter changes, including reduced fractional anisotropy on DTI
- PET/SPECT:
- Reduced striatal glucose metabolism (FDG-PET)
- Decreased dopamine D2 receptor binding (raclopride PET)
- Functional MRI:
- Altered activation patterns in task-based and resting-state studies
- Quantitative imaging:
- Volumetric analysis to track disease progression
- MR spectroscopy shows reduced N-acetylaspartate in the striatum
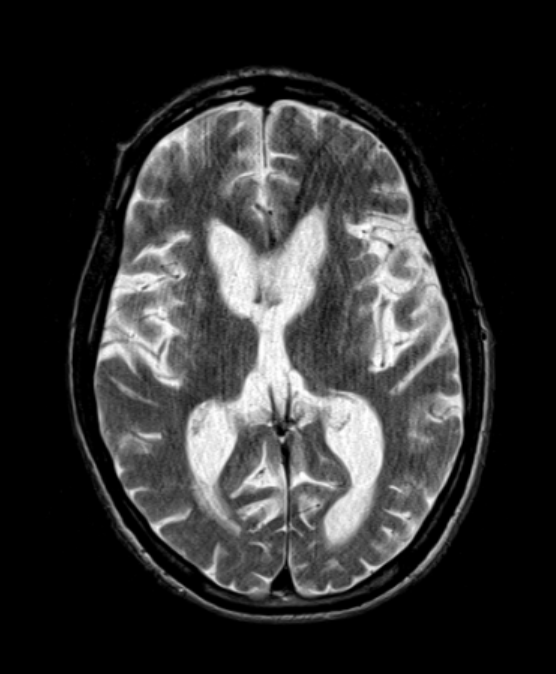
- A 50-year-old male presented with increasing clumsiness, jerky arm movements and mood swings.
- MRI showed relatively mild caudate head atrophy and putaminal T2-hyperintensity.
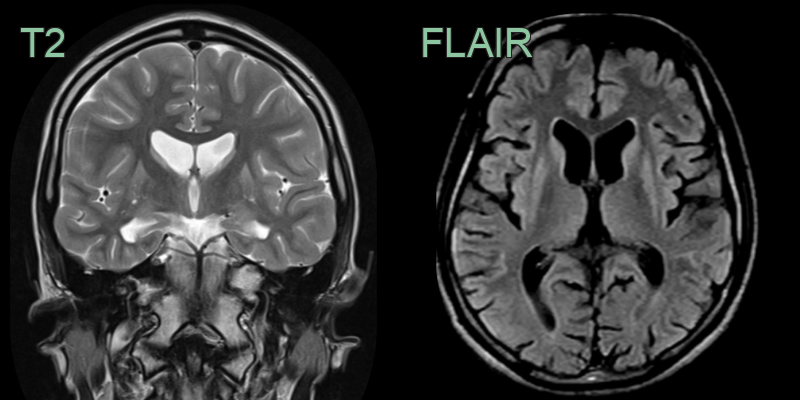
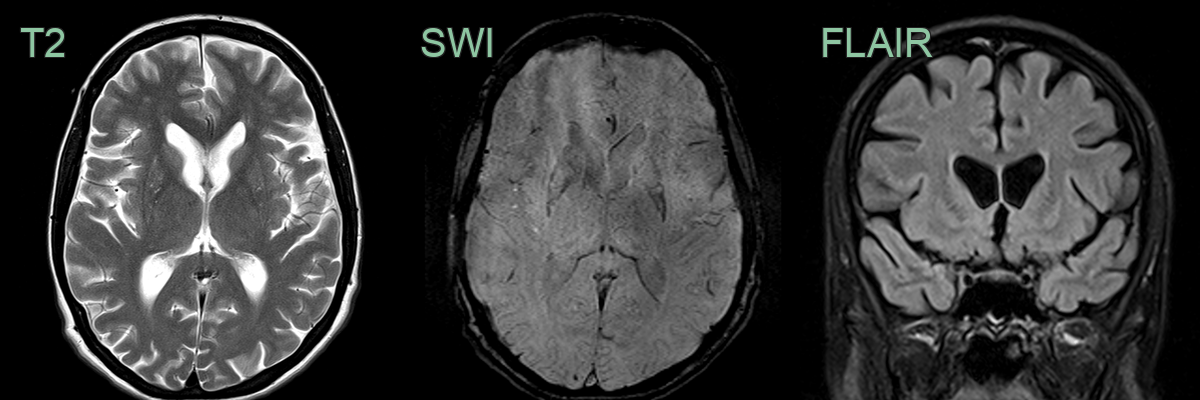
- 50-year-old male with increasing clumsiness and jerky arm movements.
- MRI showed atrophy and hyperintensity of the corpora striata on both sides.
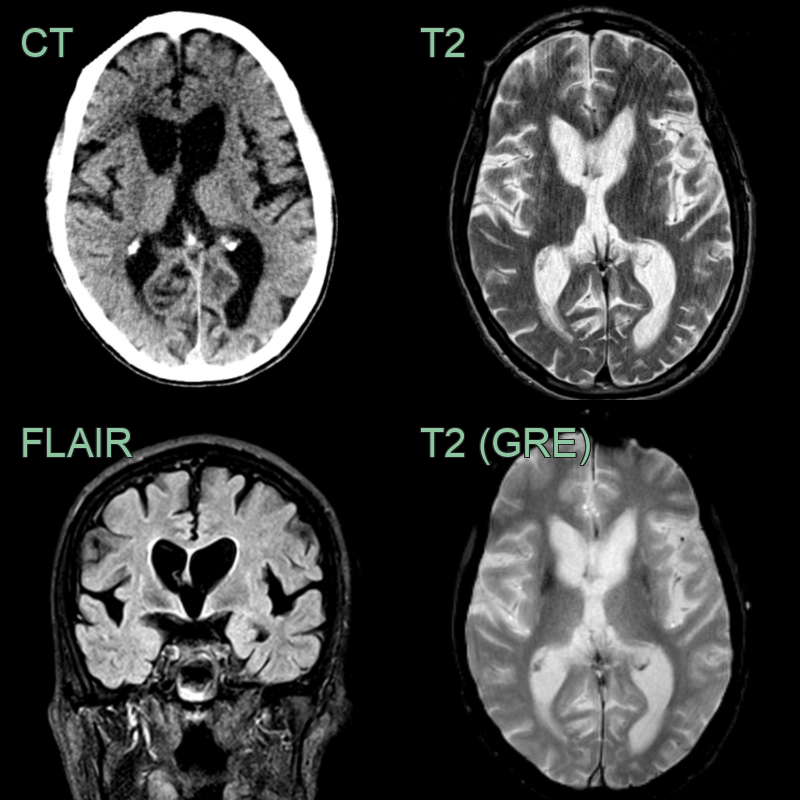
- A 55-year-old patient with a history of depression presented after worsening incoordination.
- Imaging showed marked atrophy of the caudate head. There was less pronounced atrophy of the T2-hyperintense putamina.
Treatment¶
- No cure available; management focuses on symptom control
- Pharmacological interventions:
- Tetrabenazine for chorea (FDA-approved)
- Antipsychotics for psychiatric symptoms and chorea
- Antidepressants for mood disorders
- Non-pharmacological approaches:
- Physical therapy for motor symptoms
- Occupational therapy for daily living activities
- Speech therapy for communication and swallowing difficulties
- Genetic counseling for at-risk individuals and families
- Emerging therapies:
- Gene silencing approaches (e.g., antisense oligonucleotides)
- Cell replacement strategies
- Small molecule therapies targeting mutant huntingtin
Differential diagnosis¶
| Differential Diagnosis | Distinguishing Feature |
|---|---|
| Wilson's disease | Presence of Kayser-Fleischer rings; abnormal copper metabolism |
| Spinocerebellar ataxia | Prominent cerebellar signs; different genetic mutation |
| Neuroacanthocytosis | Presence of acanthocytes on blood smear; orofacial dyskinesia |
| Frontotemporal lobar degeneration | More frontal or temporal lobar atrophy |