L-2 hydroxyglutaric Aciduria¶
Summary
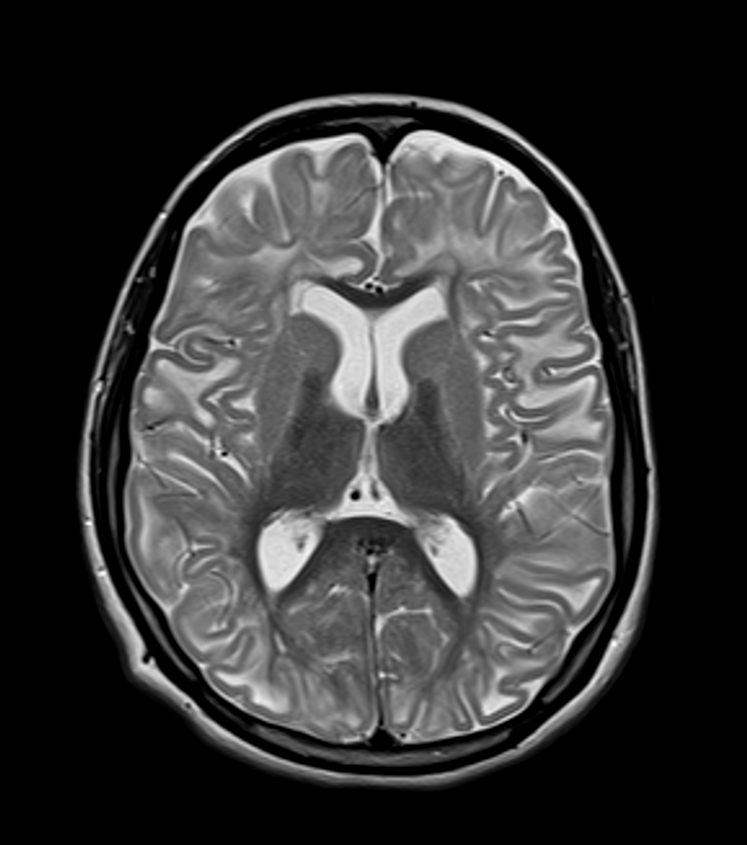
- Rare neurometabolic disorder characterised by elevated levels of L-2-hydroxyglutaric acid in urine, plasma, and cerebrospinal fluid
- Presents with developmental delay, epilepsy, and progressive neurological deterioration
- MRI shows distinctive white matter abnormalities and subcortical cystic lesions
Pathophysiology¶
- Autosomal recessive disorder caused by mutations in the L2HGDH gene
- Deficiency of L-2-hydroxyglutarate dehydrogenase enzyme leads to accumulation of L-2-hydroxyglutaric acid
- Toxic accumulation results in:
- Oxidative stress
- Mitochondrial dysfunction
- Impaired energy metabolism in the brain
Demographics¶
- Rare disorder with an estimated prevalence of <1/1,000,000
- No significant gender predilection
- Typically presents in infancy or early childhood
- Higher prevalence in populations with consanguineous marriages
Diagnosis¶
- Clinical features:
- Developmental delay
- Seizures
- Ataxia
- Macrocephaly
- Progressive cognitive decline
- Laboratory findings:
- Elevated L-2-hydroxyglutaric acid in urine, plasma, and CSF
- Genetic testing for L2HGDH mutations
- Neuroimaging findings (crucial for diagnosis)
Imaging¶
- MRI is the modality of choice
- Characteristic findings:
- Subcortical white matter abnormalities (high T2, low T1 signal)
- Sparing of deep white matter and corpus callosum
- Bilateral symmetrical involvement of:
- Cerebral hemispheres
- Basal ganglia (especially putamen and caudate nucleus)
- Dentate nuclei
- Subcortical cystic lesions, particularly in frontal and temporal lobes
- Cerebellar atrophy (late finding)
- Differential diagnosis:
- Canavan disease
- Alexander disease
- Megalencephalic leukoencephalopathy with subcortical cysts
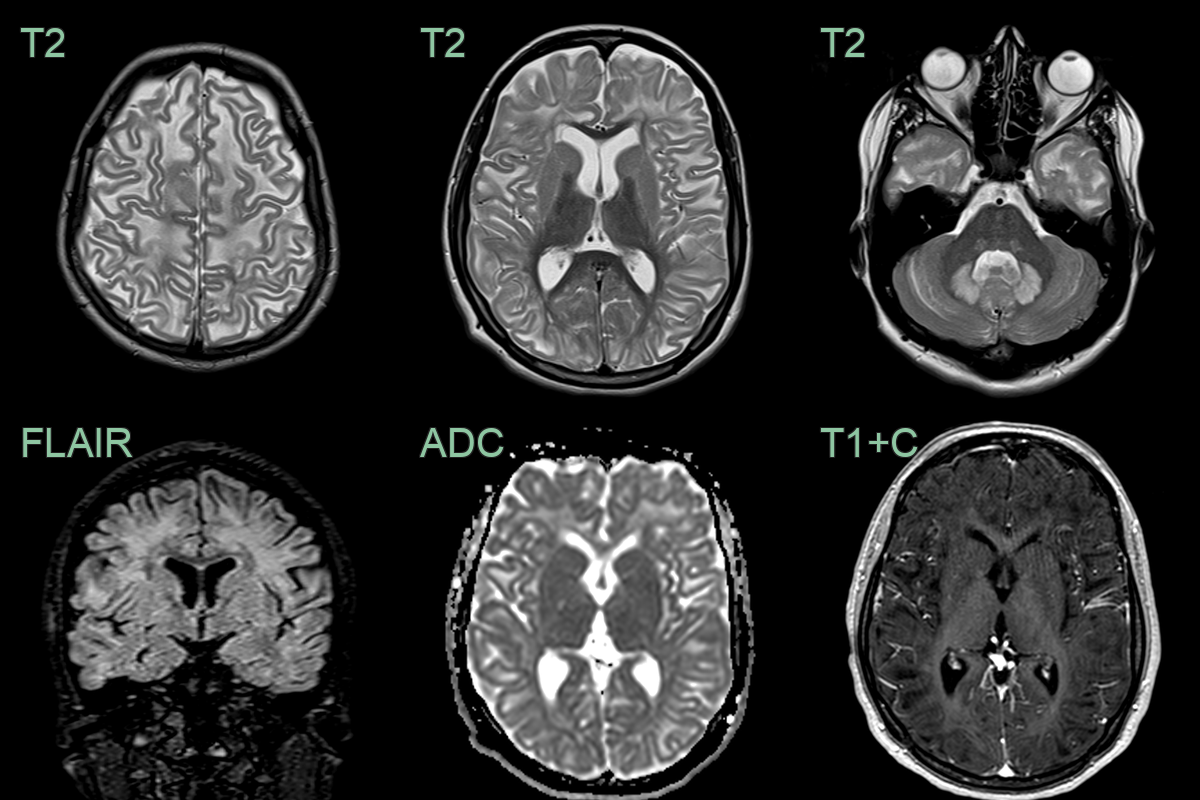
- 20-year-old patient with learning difficulties.
- MRI showed a diffuse supratentorial leukocencpehalopathy and dentate nucleus hyperintensity without diffusion restriction, significant volume loss, or pathological enhancement.
Treatment¶
- No curative treatment available
- Management is supportive and symptomatic:
- Antiepileptic drugs for seizure control
- Physical and occupational therapy
- Special education and cognitive support
- Experimental treatments under investigation:
- Riboflavin supplementation
- Triheptanoin (anaplerotic therapy)
- Genetic counselling for affected families
- Prenatal diagnosis possible for at-risk pregnancies
Differential diagnosis¶
| Differential Diagnosis | Differentiating Feature |
|---|---|
| Canavan disease | Elevated N-acetylaspartic acid in urine and CSF; macrocephaly more prominent |
| Alexander disease | Predominant frontal white matter involvement; presence of Rosenthal fibres on histology |
| Megalencephalic leukoencephalopathy with subcortical cysts | Presence of subcortical cysts; less severe clinical course |
| Glutaric aciduria type 1 | Frontotemporal atrophy; basal ganglia lesions; elevated glutaric acid in urine |
| Mitochondrial disorders | Lactate elevation; involvement of brainstem and basal ganglia |
| Sjögren-Larsson syndrome | Ichthyosis; spastic diplegia; retinal changes |
| Van der Knaap disease | Swollen white matter with cystic degeneration; milder clinical course |
| Aicardi-Goutières syndrome | Calcifications in basal ganglia; elevated interferon-alpha in CSF |
| Metachromatic leukodystrophy | Peripheral neuropathy; sulfatide accumulation in urine |
| Krabbe disease | Peripheral neuropathy; elevated galactocerebrosidase levels |