Labrune Syndrome¶
Summary
- Rare genetic disorder characterised by leukoencephalopathy, brain calcifications, and cysts
- Caused by mutations in the SNORD118 gene, affecting small nucleolar RNA function
- Typically presents in childhood with neurological symptoms and progressive course
Pathophysiology¶
- Autosomal recessive inheritance pattern
- Mutations in SNORD118 gene, encoding U8 small nucleolar RNA
- Disruption of ribosomal RNA processing and protein synthesis
- Leads to white matter abnormalities, calcifications, and cyst formation in the brain
- Exact mechanism of cyst formation remains unclear
Demographics¶
- Rare disorder with fewer than 50 cases reported worldwide
- No clear gender predilection
- Typically presents in childhood, but adult-onset cases have been described
- Most reported cases are from consanguineous families
Diagnosis¶
- Clinical presentation:
- Developmental delay or regression
- Seizures
- Ataxia
- Spasticity
- Cognitive decline
- Genetic testing:
- Identification of biallelic mutations in SNORD118 gene
- Neuroimaging findings (see Imaging section)
- Exclusion of other leukoencephalopathies and calcifying disorders
Imaging¶
- Computed Tomography (CT):
- Bilateral calcifications in basal ganglia, thalami, and subcortical white matter
- Hypodense cystic lesions
- Magnetic Resonance Imaging (MRI):
- T2-weighted and FLAIR hyperintensities in cerebral white matter
- Cystic lesions with variable signal intensity
- Calcifications appear as hypointense foci on T2* or susceptibility-weighted imaging
- MR Spectroscopy:
- Reduced N-acetylaspartate (NAA) peak
- Elevated choline and lactate peaks in affected white matter
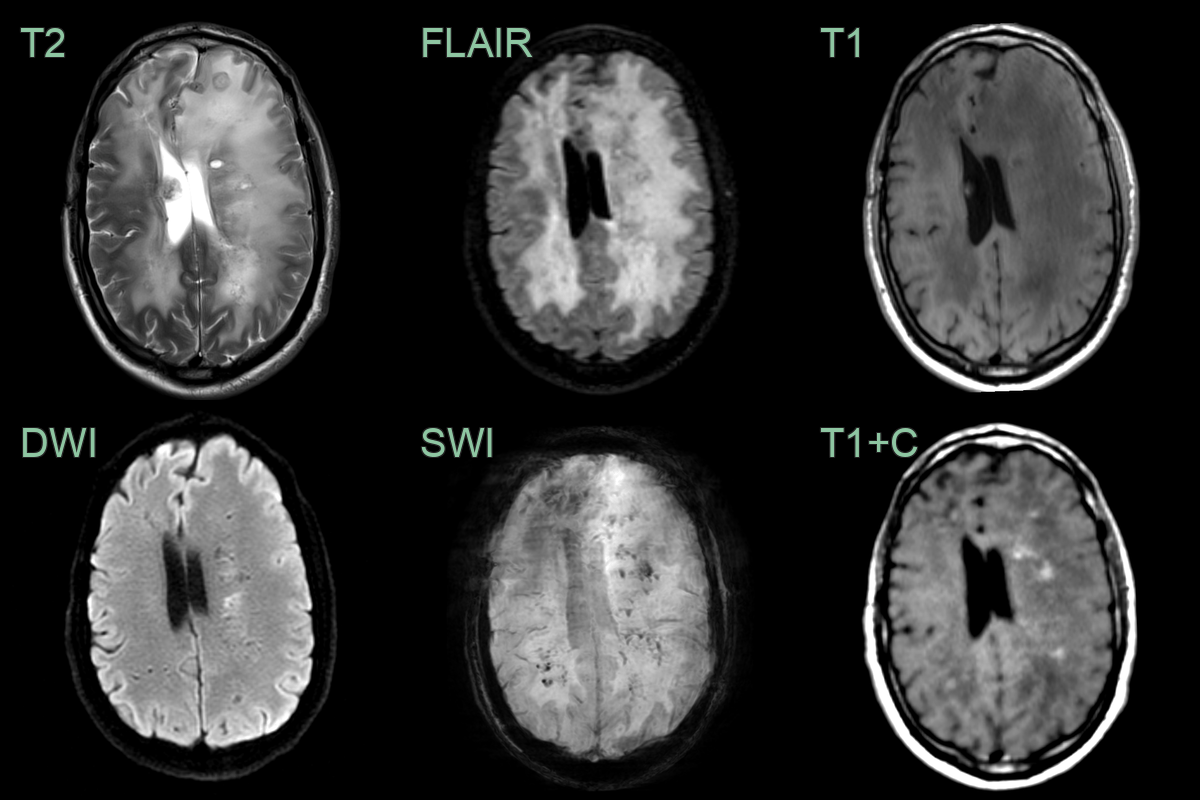
- A 30-year-old patient with known diagnosis of Labrune syndrome had a surveillence scan after surgery to drain a cyst (no shown).
- MRI showed a diffuse leukoencephalopathy within microcalcification and punctate enhancement.
Treatment¶
- No curative treatment available
- Management is supportive and symptomatic:
- Anticonvulsants for seizure control
- Physical therapy for motor symptoms
- Occupational therapy for daily living activities
- Speech therapy for language difficulties
- Regular neurological and developmental assessments
- Genetic counselling for affected families
- Ongoing research into potential gene therapy approaches
Differential diagnosis¶
| Differential Diagnosis | Differentiating Feature |
|---|---|
| Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) | Absence of calcifications on brain imaging |
| Coats plus syndrome | Presence of retinal telangiectasias and exudates |
| Aicardi-Goutières syndrome | Earlier onset, typically in infancy |
| Primary familial brain calcification (Fahr's disease) | Lack of white matter abnormalities |
| Cerebroretinal microangiopathy with calcifications and cysts (CRMCC) | Presence of retinal vascular abnormalities |
| Mitochondrial encephalopathy | Typically associated with other systemic manifestations |
| Multiple sclerosis | Absence of calcifications and cysts on brain imaging |
| Cerebral amyloid angiopathy | Typically affects older adults, lacks cysts |
| Cerebral vasculitis | Absence of calcifications, different pattern of white matter changes |
| Leukoencephalopathy with calcifications and cysts (LCC) | Considered the same entity as Labrune syndrome by some researchers |