Leigh Disease¶
Summary
- Rare, progressive neurometabolic disorder characterised by bilateral symmetric lesions in the basal ganglia, thalami, and brainstem
- Caused by mutations affecting mitochondrial energy production
- Typically presents in infancy with developmental delay, seizures, and lactic acidosis
Pathophysiology¶
- Genetic mutations affecting mitochondrial energy production, particularly in the respiratory chain complexes
- Deficiency in pyruvate dehydrogenase complex or coenzyme Q10
- Results in cellular energy failure, particularly affecting high-energy demand tissues like the brain
- Neuronal loss and demyelination in affected areas
Demographics¶
- Incidence: approximately 1 in 40,000 live births
- Typically presents in infancy or early childhood
- Can rarely present in adolescence or adulthood
- No significant gender predilection
- Higher prevalence in certain populations (e.g., Saguenay-Lac-Saint-Jean region of Quebec)
Diagnosis¶
- Clinical presentation:
- Developmental delay or regression
- Seizures
- Ataxia
- Dystonia
- Respiratory difficulties
- Laboratory findings:
- Elevated lactate in blood and/or cerebrospinal fluid
- Pyruvate elevation
- Abnormal respiratory chain enzyme activities
- Genetic testing:
- Mitochondrial DNA mutations
- Nuclear DNA mutations affecting mitochondrial function
- Muscle biopsy:
- May show ragged red fibres or cytochrome c oxidase-negative fibres
Imaging¶
- MRI:
- Bilateral, symmetric T2 hyperintensities in:
- Basal ganglia (particularly putamen)
- Thalami
- Brainstem (particularly midbrain and pons)
- Cerebral and cerebellar white matter involvement may occur
- Contrast enhancement is uncommon
- CT:
- May show hypodensities in affected areas
- Less sensitive than MRI
- MR Spectroscopy:
- Elevated lactate peak at 1.3 ppm
- Decreased N-acetylaspartate (NAA) peak
- Diffusion-weighted imaging:
- May show restricted diffusion in acute lesions
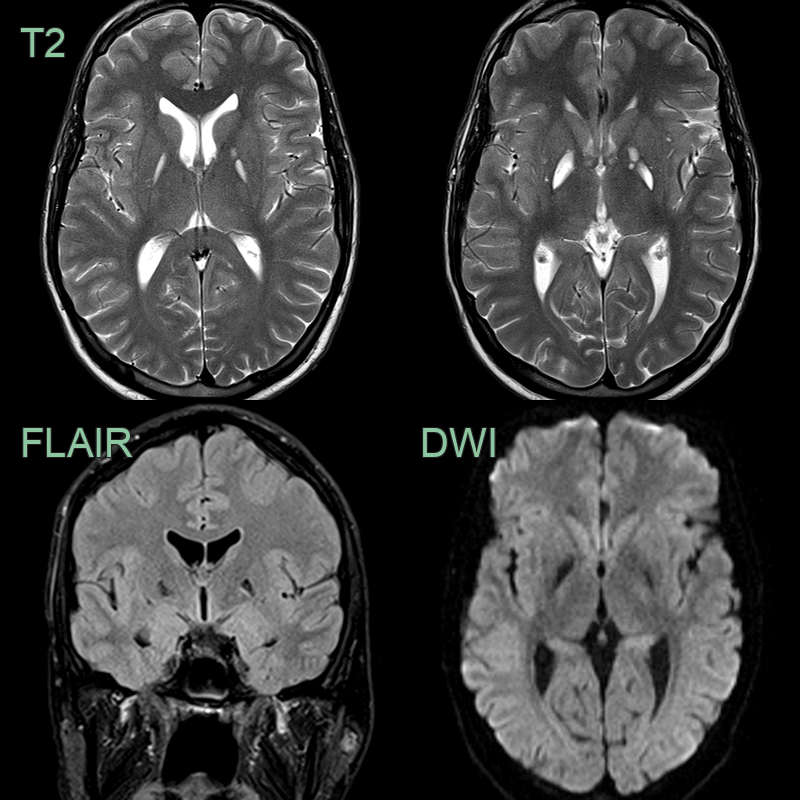
- A 25-year-old patient presented following a slightly worsening of longstanding spastic paresis and dysarthria.
- MRI showed old damage in the basal ganglia without DWI hyperintensity.
- Genomic testing revealed X-linked Leigh disease caused by a hemizygous mutation in the PDHA1 gene.
Treatment¶
- No curative treatment available
- Supportive care and symptom management:
- Anticonvulsants for seizure control
- Respiratory support as needed
- Nutritional support
- Mitochondrial cocktail:
- Coenzyme Q10
- Thiamine
- Riboflavin
- L-carnitine
- Alpha-lipoic acid
- Dichloroacetate:
- May help reduce lactic acidosis
- Ketogenic diet:
- May be beneficial in some cases
- Gene therapy:
- Experimental approaches under investigation
- Prognosis:
- Generally poor, with most patients not surviving beyond early childhood
- Some individuals with milder variants may have longer survival
Differential diagnosis¶
| Differential Diagnosis | Distinguishing Feature |
|---|---|
| Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) | Stroke-like episodes and migraines are more common in MELAS; Leigh disease typically presents earlier in life |
| Pyruvate dehydrogenase deficiency | Often presents with congenital lactic acidosis; Leigh disease typically has later onset |
| Biotinidase deficiency | Skin rash and alopecia are common; not typically seen in Leigh disease |
| Neurodegeneration with brain iron accumulation | Iron accumulation in basal ganglia on MRI; not typically seen in Leigh disease |