Linear Scleroderma¶
Summary
- Linear scleroderma is a localised form of scleroderma characterised by fibrosis and inflammation of the skin and underlying tissues in a linear distribution
- Typically affects children and young adults, with a female predominance
- Diagnosis is based on clinical presentation, with imaging playing a supportive role in assessing extent and complications
Pathophysiology¶
- Autoimmune disorder with unclear etiology
- Characterised by excessive collagen deposition and fibrosis in affected areas
- Involves skin, subcutaneous tissue, and may extend to underlying muscle and bone
- Vascular changes and inflammation precede fibrosis
Demographics¶
- Onset typically in childhood or young adulthood
- Female to male ratio of approximately 2.5:1
- Incidence estimated at 0.4-2.7 per 100,000 person-years
- More common in Caucasians, but can affect all racial groups
Diagnosis¶
- Primarily based on clinical presentation and physical examination
- Characteristic linear distribution of skin changes
- Skin biopsy may be performed to confirm diagnosis
- Laboratory tests (ANA, anti-scl-70) often negative or non-specific
- Differential diagnosis includes morphea, en coup de sabre, and Parry-Romberg syndrome
Imaging¶
-
Ultrasound:
- Increased dermal thickness and echogenicity
- Reduced subcutaneous tissue
- Useful for monitoring disease progression and treatment response
-
MRI:
- T1-weighted images: Hypointense signal in affected areas
- T2-weighted images: Hyperintense signal in active lesions
- Contrast-enhanced: Enhancement in active lesions
- Useful for assessing deep tissue involvement and monitoring disease activity
-
X-ray:
- May show soft tissue atrophy and underlying bone involvement
- Useful for detecting growth disturbances in paediatric patients
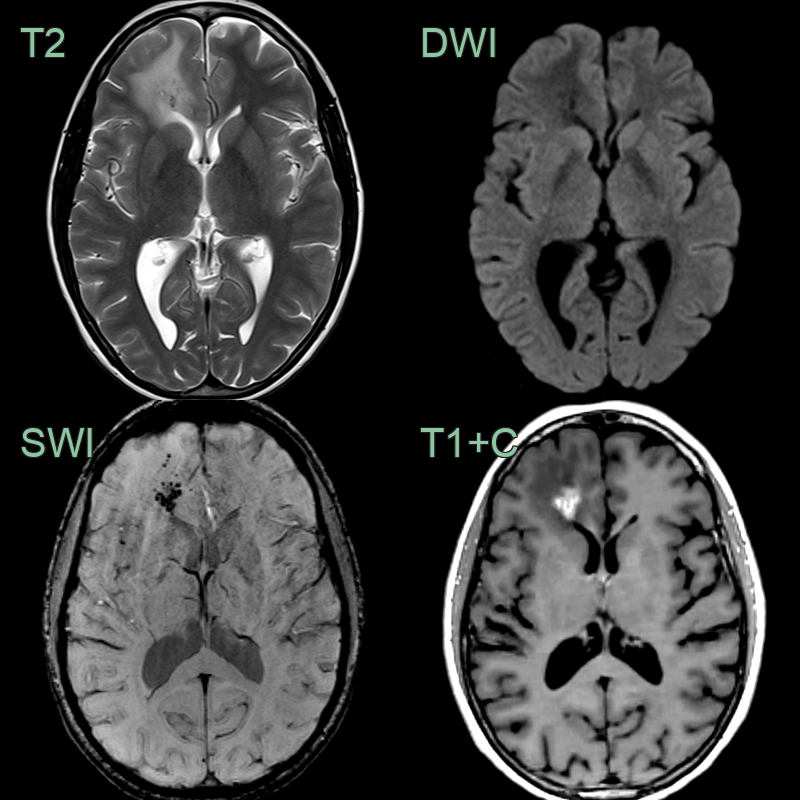
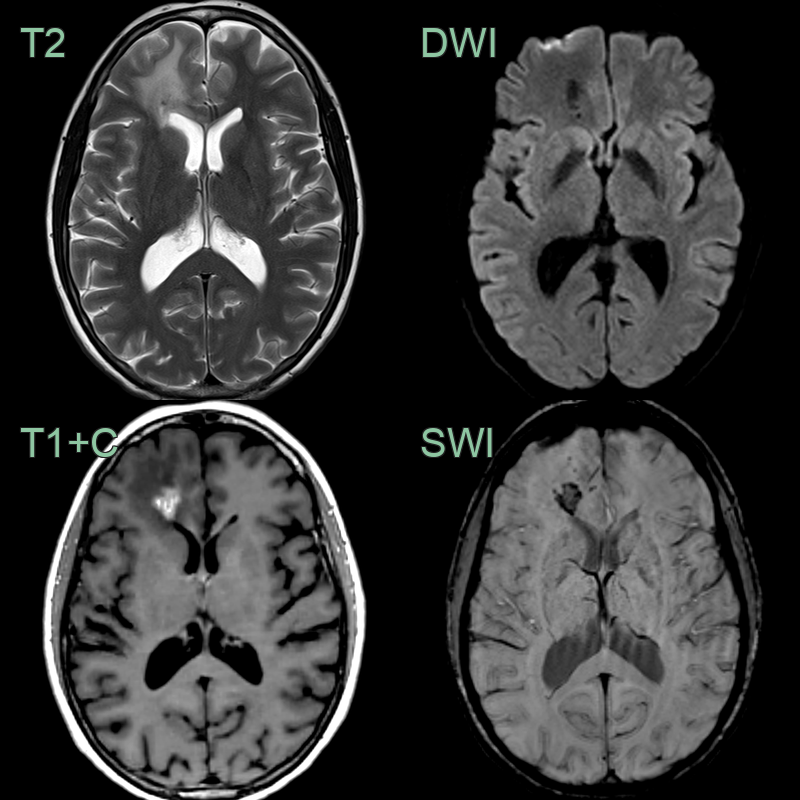
- 45-year-old patient had an incidental lesion in the right frontal lobe.
- Thinning of the subcutaneous tissue over the forehead represented an en coup de sabre (red arrow).
- MRI showed an enhancing lesion with siderosis and subtle calcicification.
- Enhancement within the lesion decreased (without treatment) to leave a peripherally enhancing cavity.
Treatment¶
- Multidisciplinary approach involving rheumatologists, dermatologists, and physical therapists
- Topical treatments:
- Corticosteroids
- Calcineurin inhibitors (tacrolimus, pimecrolimus)
- Systemic treatments:
- Methotrexate (first-line systemic therapy)
- Mycophenolate mofetil
- Systemic corticosteroids (for rapidly progressive disease)
- Phototherapy:
- UVA1 phototherapy
- Narrowband UVB
- Physical therapy and rehabilitation to maintain range of motion and prevent contractures
- Regular monitoring for disease progression and complications
Differential diagnosis¶
| Differential diagnosis | Differentiating feature |
|---|---|
| Sturge-Weber syndrome | Leptomeningeal angiomatosis; cortical calcification ("tram-track"); ipsilateral cortical atrophy; facial port-wine stain; no dermal fibrosis |
| Rasmussen encephalitis | Progressive unilateral cortical and subcortical atrophy; T2 signal change; inflammatory; associated with drug-resistant epilepsy |
| Focal cortical dysplasia | Cortical thickening; blurring of the grey-white junction; transmantle sign on FLAIR; no enhancement or haemosiderin |
| Cavernous malformation | "Popcorn" T2* susceptibility appearance; complete haemosiderin rim; no progressive cortical atrophy |
| Low-grade glioma | Infiltrative T2/FLAIR hyperintensity; minimal or no enhancement; mass effect; no siderosis or cortical tethering |
| Meningioangiomatosis | Cortical-based calcified lesion; meningeal-cortical fusion; young patient; can mimic cortical dysplasia |
| Hemimegalencephaly | Unilateral cerebral enlargement; ipsilateral cortical malformation; dysmorphic enlarged ventricle; no atrophy |