Mitochondrial Encephalopathy with Lactic Acidosis and Stroke-like Episodes (MELAS)¶
Summary
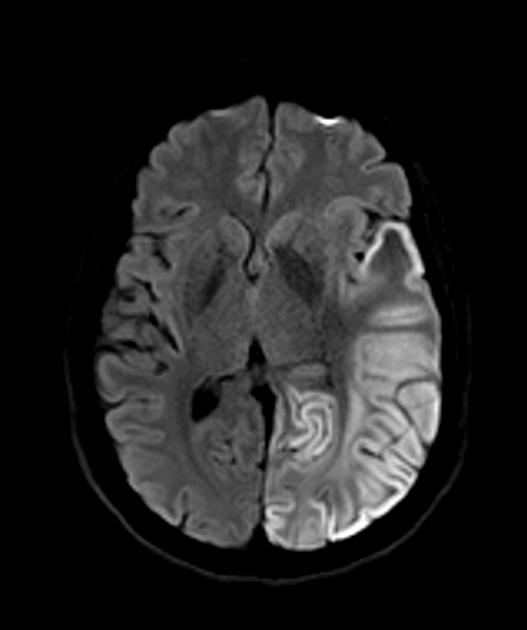
- MELAS is a rare mitochondrial disorder characterised by encephalopathy, lactic acidosis, and stroke-like episodes
- Caused by mutations in mitochondrial DNA, most commonly in the MT-TL1 gene
- Imaging findings include stroke-like lesions not conforming to vascular territories, often in the posterior cerebral regions
Pathophysiology¶
- Caused by mutations in mitochondrial DNA, primarily affecting energy production in cells
- Most common mutation (80% of cases) is A3243G in the MT-TL1 gene
- Impaired oxidative phosphorylation leads to:
- Cellular energy deficiency
- Increased anaerobic metabolism and lactic acid production
- Neuronal dysfunction and cell death
Demographics¶
- Rare disorder with estimated prevalence of 0.18 per 100,000
- Typically presents in childhood or early adulthood
- No significant gender predilection
- Maternal inheritance pattern due to mitochondrial DNA mutation
Diagnosis¶
- Clinical presentation:
- Stroke-like episodes
- Seizures
- Headaches
- Muscle weakness and exercise intolerance
- Hearing loss
- Diabetes mellitus
- Laboratory findings:
- Elevated serum and CSF lactate levels
- Elevated serum pyruvate levels
- Ragged-red fibres on muscle biopsy
- Genetic testing:
- Mitochondrial DNA analysis for common mutations
Imaging¶
- CT findings:
- Hypodense lesions in cortical and subcortical regions
- Calcifications in basal ganglia and cerebral white matter
- MRI findings:
- T2/FLAIR hyperintense lesions not conforming to vascular territories
- Predilection for posterior cerebral regions (occipital, parietal, and temporal lobes)
- Cortical and subcortical involvement
- Restricted diffusion in acute lesions
- Lesions may show contrast enhancement
- MR Spectroscopy:
- Elevated lactate peak at 1.3 ppm
- Decreased N-acetylaspartate (NAA) peak
- Perfusion imaging:
- Hyperperfusion in acute lesions
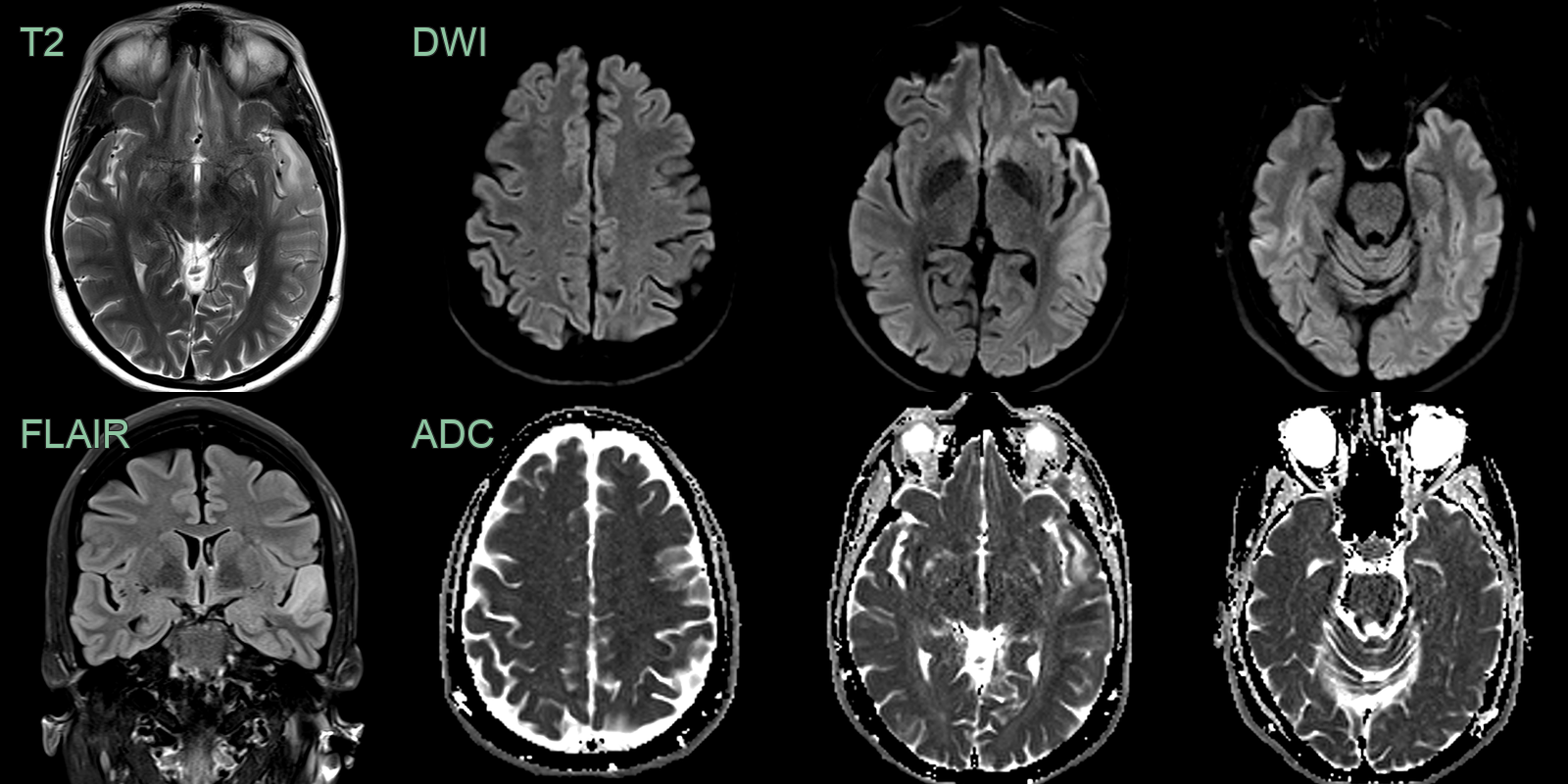
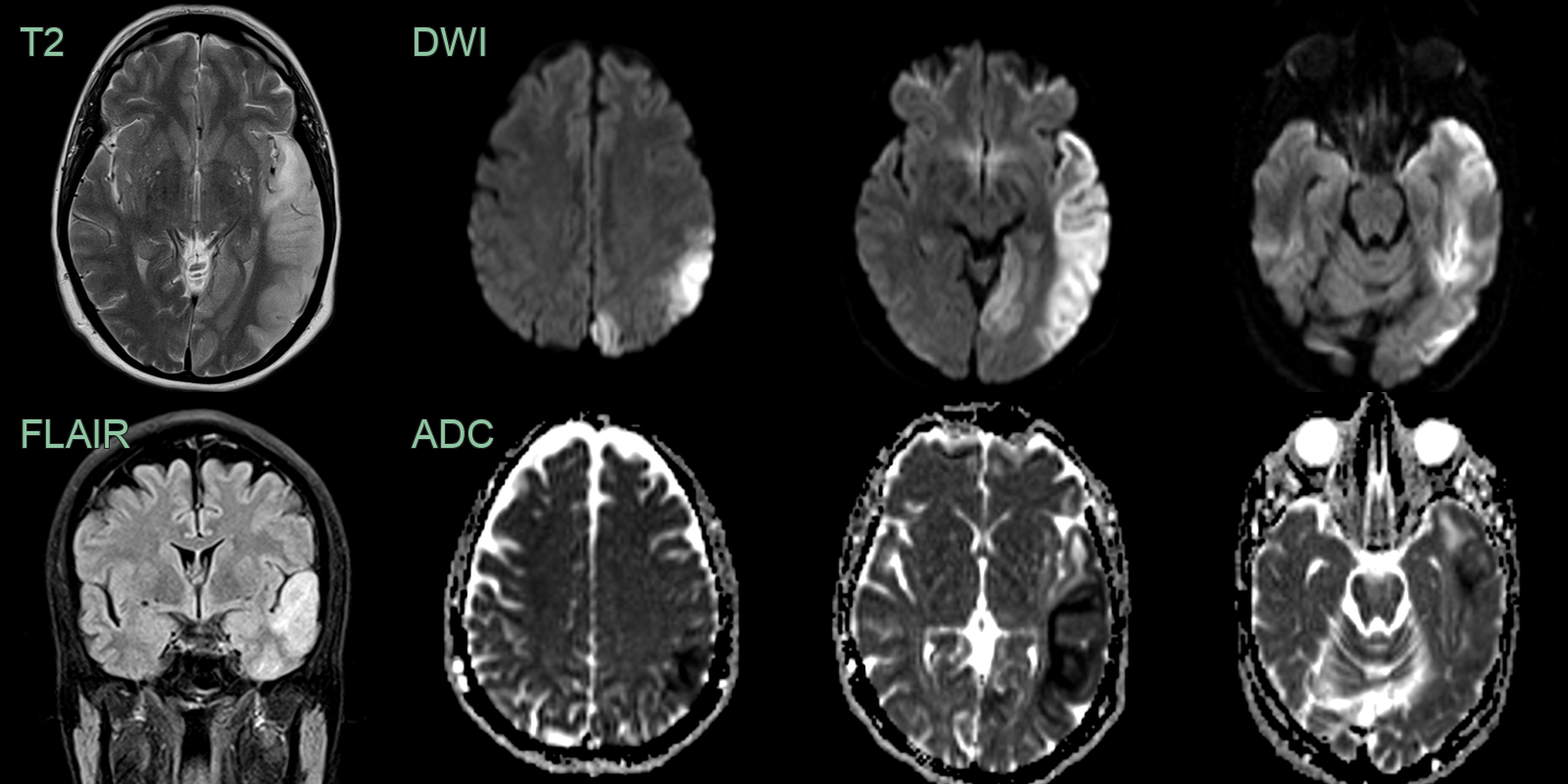
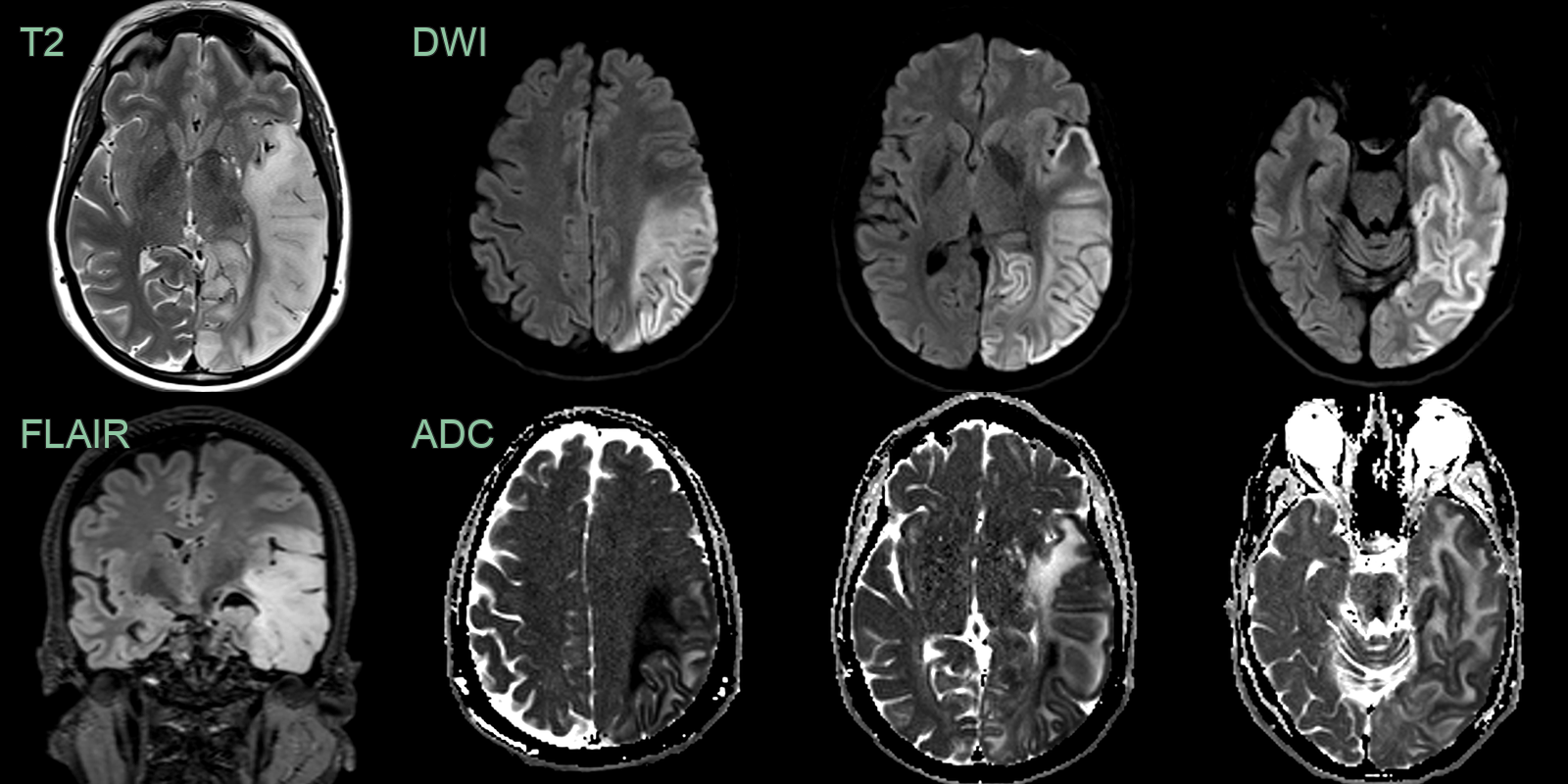
- 40-year-old patient presented with acute onset of aphasia.
- Initial MRI showed cortical and subcortical diffusion-weighted abnormality in the left anterior temporal lobe.
- On follow-up imaging, this region of signal abnormality and swelling significantly increased. There was both restricted and facilitiated diffusion involving both cortex and subcortical white matter.
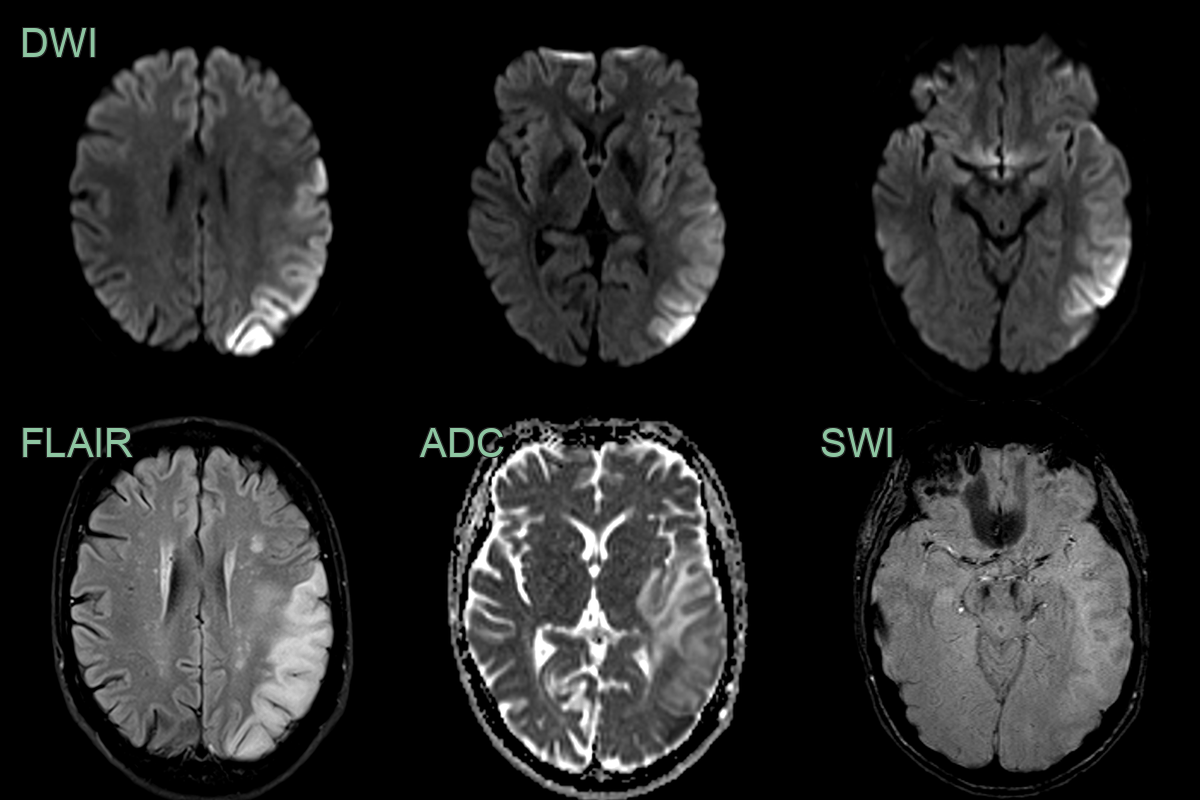
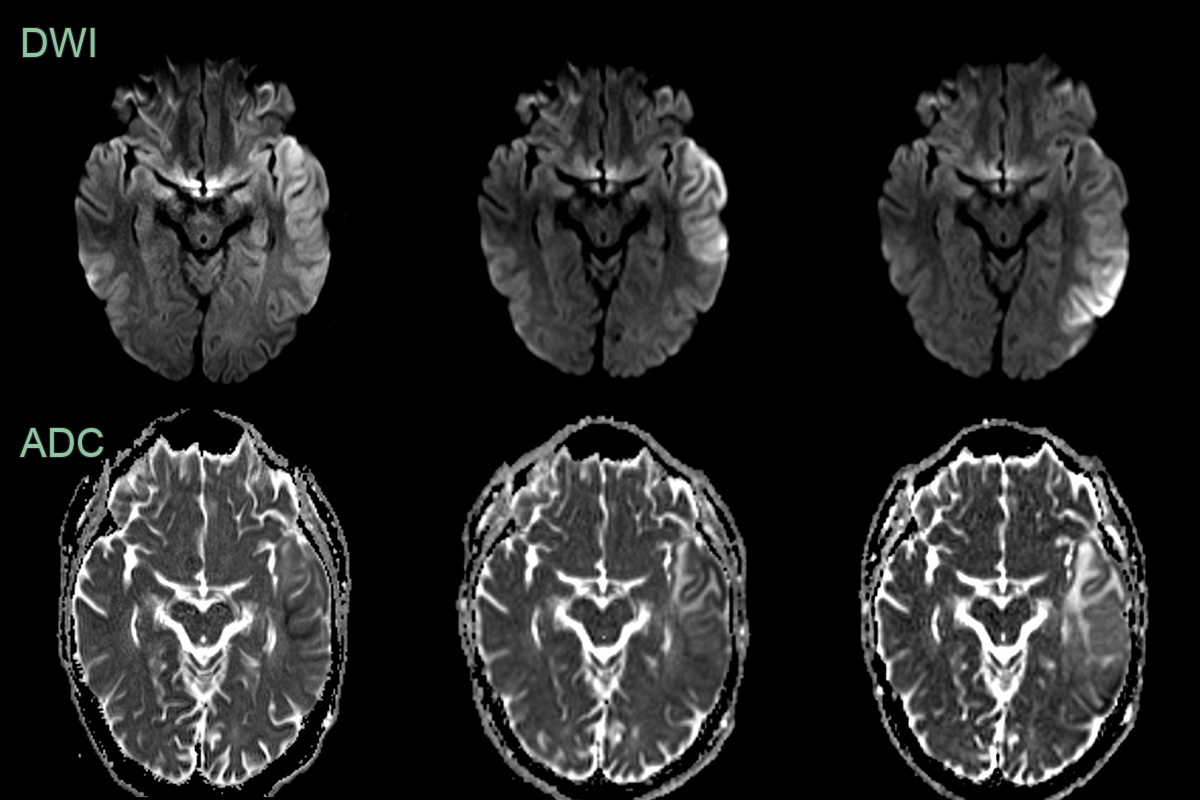
- A 30-year-old patient presented with a 1 week history of word finding difficulty followed by a seizure.
- MRI showed cortical diffusion restriction variably affecting the cortex and subcortical white matter of the left cerebral hemisphere.
- The region of abnormality increased over the 3 weeks.
Treatment¶
- No curative treatment available
- Management focuses on symptom control and supportive care:
- Anticonvulsants for seizure control
- L-arginine for acute stroke-like episodes
- Coenzyme Q10 and other antioxidants as mitochondrial supplements
- Management of diabetes mellitus and other systemic manifestations
- Genetic counselling for affected families
- Avoid valproic acid due to risk of liver failure in mitochondrial disorders
- Regular monitoring of cardiac, renal, and endocrine function
- Physical and occupational therapy for muscle weakness and coordination issues
Differential diagnosis¶
| Differential Diagnosis | Distinguishing Feature |
|---|---|
| Stroke | Typically affects older patients; vascular territories respected; no lactic acidosis |
| Multiple Sclerosis | Periventricular white matter lesions; no lactic acidosis; oligoclonal bands in CSF |
| Encephalitis | Fever; CSF abnormalities; infectious etiology often identified |
| Leigh Syndrome | Typically affects infants; basal ganglia involvement; brainstem lesions |
| Wernicke's Encephalopathy | History of alcoholism or malnutrition; thiamine deficiency; mammillary body involvement |
| Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) | Family history; white matter lesions; no lactic acidosis |
| Migraine with aura | Reversible symptoms; no persistent imaging abnormalities; no lactic acidosis |
| Posterior Reversible Encephalopathy Syndrome (PRES) | Associated with hypertension; typically reversible; parieto-occipital predilection |
| Brain tumour | Mass effect; contrast enhancement; no lactic acidosis (unless high-grade) |
| Hashimoto's encephalopathy | Thyroid antibodies present; responsive to steroids; no lactic acidosis |