MOGAD¶
Summary
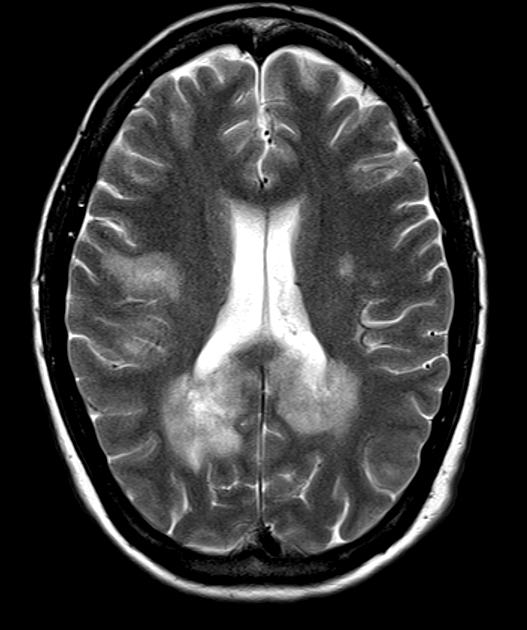
- MOGAD (Myelin Oligodendrocyte Glycoprotein Antibody-associated Disease) is an autoimmune demyelinating CNS disorder caused by antibodies against myelin oligodendrocyte glycoprotein (MOG-IgG)
- Presents with optic neuritis, acute disseminated encephalomyelitis (ADEM), transverse myelitis, or brainstem encephalitis
- Distinct from multiple sclerosis and NMOSD; generally more steroid-responsive with better prognosis
Pathophysiology¶
- Pathogenic IgG1 antibodies target MOG, a glycoprotein expressed on the outer surface of the myelin sheath and oligodendrocyte cell membrane
- MOG-IgG activates complement and antibody-dependent cellular cytotoxicity, causing demyelination
- Can be monophasic or relapsing; relapsing course more common in adults
- Distinct from aquaporin-4 (AQP4)-IgG associated NMOSD, though phenotypic overlap exists
Demographics¶
- Affects all age groups; bimodal distribution with peaks in childhood and adulthood
- Childhood cases more likely to present as ADEM
- No significant sex predilection in children; slight male predominance in adults
- Prevalence estimated at 1–2 per 100,000
Diagnosis¶
- Clinical syndromes:
- Optic neuritis: often bilateral, with disc swelling and good visual recovery
- ADEM-like: multifocal, often with encephalopathy (especially in children)
- Transverse myelitis: often short segment, conus involvement common
- Brainstem encephalitis: area postrema syndrome less common than in AQP4-NMOSD
- Laboratory:
- MOG-IgG positive in serum (cell-based assay preferred)
- CSF: lymphocytic pleocytosis in acute attacks; oligoclonal bands typically absent
- Diagnosis requires compatible clinical syndrome plus seropositivity for MOG-IgG
Imaging¶
- MRI Brain:
- T2/FLAIR: cortical/subcortical and deep white matter lesions; often large and fluffy
- ADEM pattern: bilateral, asymmetric, poorly marginated T2-hyperintense lesions
- Cortical lesions: more common than in MS or AQP4-NMOSD
- T1+C: variable enhancement; leptomeningeal enhancement may be seen
- MRI Optic Nerves:
- T2: hyperintensity along optic nerve; often anterior with perineural enhancement
- T1+C: enhancement of optic nerve sheath complex; bilateral involvement common
- MRI Spine:
- T2: conus medullaris involvement; central cord signal change; typically short segment
- Longitudinally extensive transverse myelitis can occur (≥3 segments)
- T1+C: patchy or diffuse cord enhancement
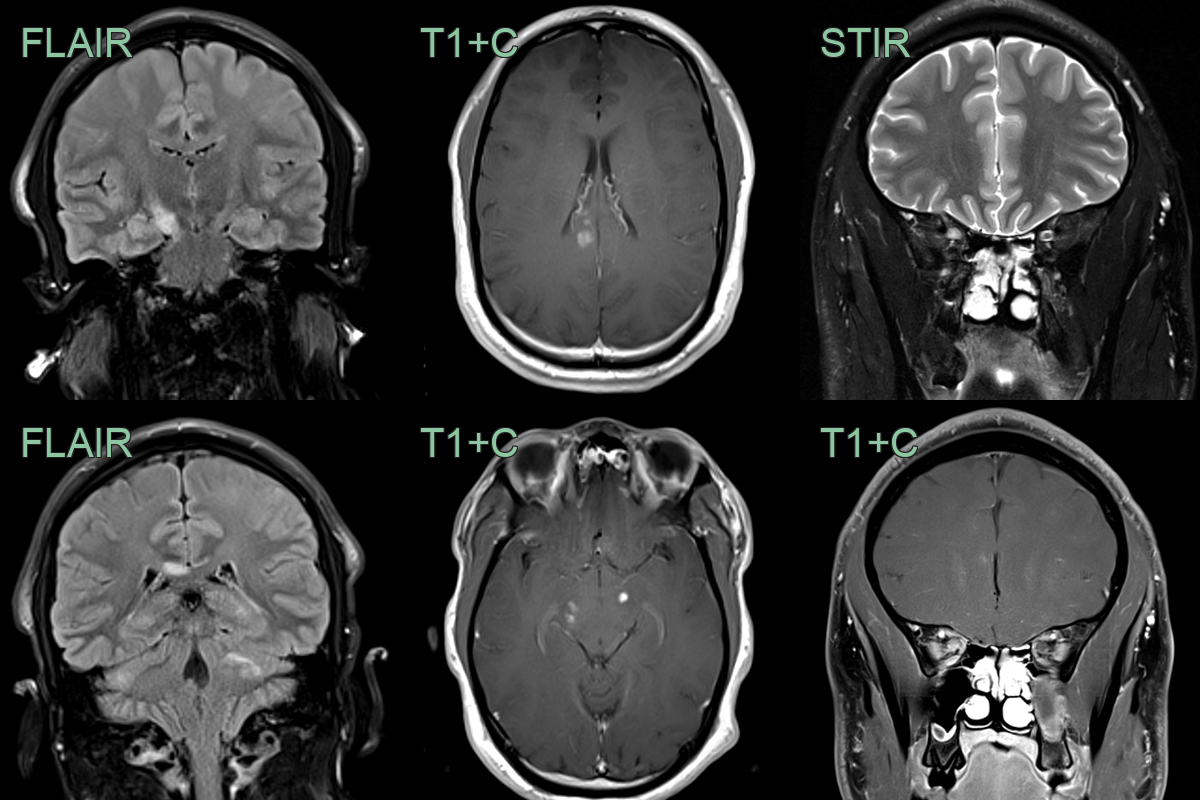
- A 30-year-old patient presented with ataxia and optic neuritis.
- MRI showed multiple enhancing lesions in the cerebellum, cerebral peduncles, corpus callosum and intraorbital right optic nerve.
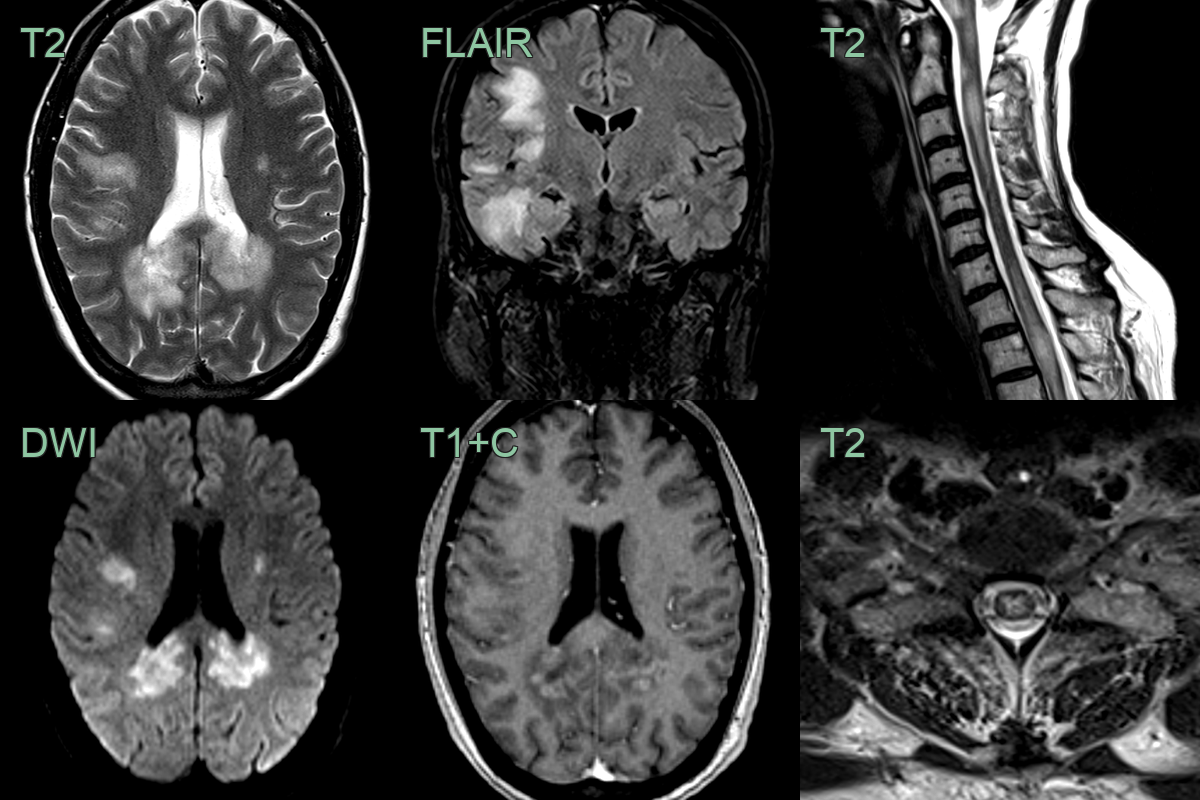
- A 40 year old presented with confusion and upper and lower limb weakness and sensory disturbance.
- MRI showed extensive white matter lesions within both cerebral hemispheres associated with diffusion restriction and peripheral enhancement.
- In the cord, there were multiple swollen mainly central short segment cord lesions.
Treatment¶
- Acute attack:
- High-dose IV methylprednisolone (1g/day for 3–5 days) — highly responsive
- IV immunoglobulin (IVIG) for steroid-refractory attacks or in children
- Plasma exchange for severe or steroid-resistant attacks
- Maintenance (relapsing disease):
- Prolonged low-dose oral prednisolone (many patients relapse on steroid taper)
- Azathioprine or mycophenolate mofetil
- IVIG (regular infusions)
- Rituximab for refractory cases
- Monitoring:
- MOG-IgG titres may correlate with disease activity
- Regular MRI and visual acuity monitoring
- Prognosis:
- Generally better than AQP4-NMOSD and MS
- Visual recovery usually good
- Relapsing course in ~50% of adults
Differential diagnosis¶
| Differential Diagnosis | Distinguishing Feature |
|---|---|
| Multiple Sclerosis | Dawson fingers and calloso-septal interface lesions; short spinal cord lesions (<2 vertebral segments); no leptomeningeal enhancement |
| Neuromyelitis Optica spectrum disorder | Area postrema and medulla lesions; posterior optic nerve and chiasm involvement; longitudinally extensive transverse myelitis |
| Acute Disseminated Encephalomyelitis | Large, confluent, bilateral T2 lesions involving grey and white matter; often involves basal ganglia and thalami |