Neurofibroma¶
Summary
- Benign peripheral nerve sheath tumour composed of Schwann cells, fibroblasts, and perineural cells
- Often associated with neurofibromatosis type 1 (NF1)
- Imaging shows well-defined, fusiform soft tissue masses along the course of peripheral nerves
Pathophysiology¶
- Arise from the connective tissue surrounding peripheral nerve sheaths
- Composed of a mixture of cell types:
- Schwann cells (predominant)
- Fibroblasts
- Perineural cells
- Mast cells
- Associated with mutations in the NF1 gene, which encodes neurofibromin, a tumour suppressor protein
- In sporadic cases, somatic mutations in the NF1 gene may occur
Demographics¶
- Can occur at any age, but most common in young adults
- No significant gender predilection
- Higher prevalence in individuals with NF1:
- Approximately 95% of NF1 patients develop multiple neurofibromas
- Sporadic cases can occur in individuals without NF1
Diagnosis¶
- Clinical presentation:
- Often asymptomatic
- May cause pain, paresthesia, or neurological deficits if compressing adjacent structures
- Physical examination:
- Palpable, soft, rubbery masses along the course of peripheral nerves
- Positive Tinel's sign (tingling sensation when tapping over the tumour)
- Genetic testing:
- NF1 gene mutation analysis in suspected cases of NF1
Imaging¶
- Ultrasound:
- Hypoechoic, well-defined fusiform masses
- Typically show posterior acoustic enhancement
- MRI:
- T1-weighted: isointense to slightly hypointense to muscle
- T2-weighted: hyperintense with central hypointense focus ("target sign")
- STIR: hyperintense
- Contrast-enhanced T1: variable enhancement patterns
- CT:
- Isodense to muscle
- May show remodeling of adjacent bone in long-standing cases
- PET/CT:
- Generally low FDG uptake (SUV < 2.5)
- Higher uptake may indicate malignant transformation
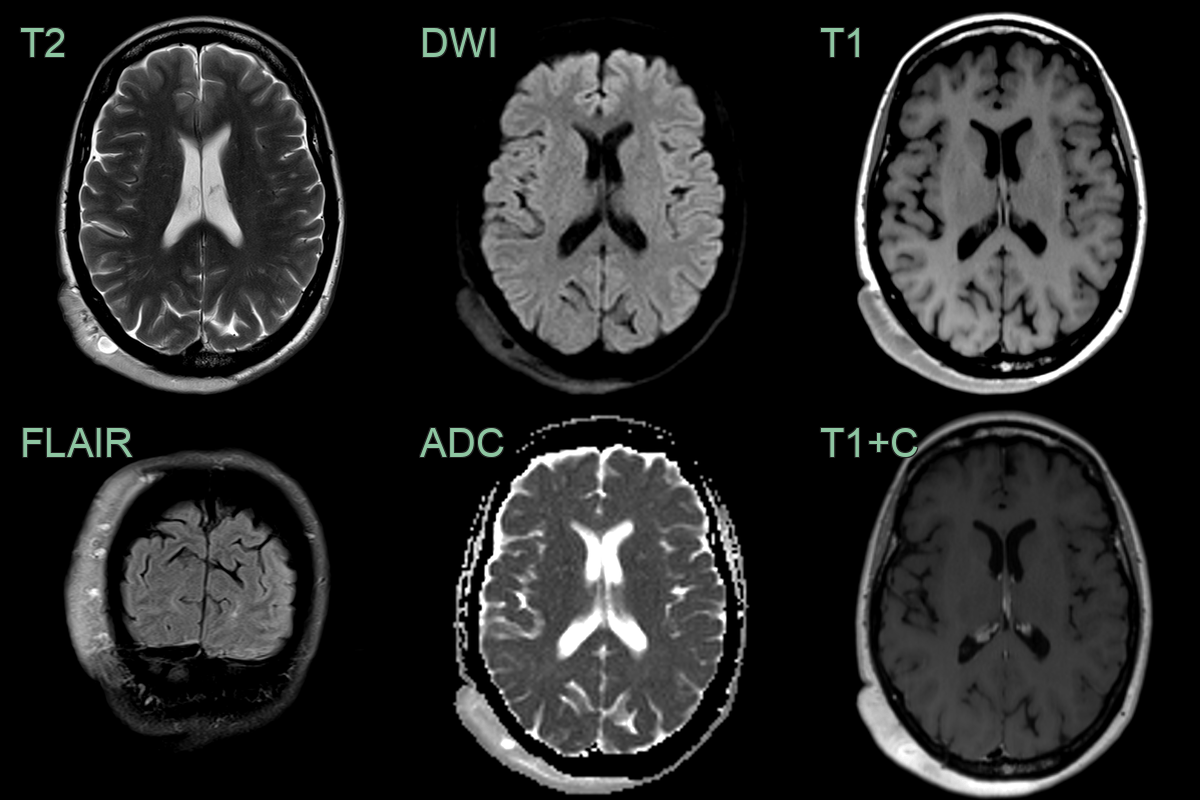
- 50-year-old patient with neurofibromatosis presented with an enlarging painless lesion over the back of the head.
- The lesion enhanced homogeneously (aside from a few cyst-like regions) and the underlying bone was normal.
- The diffuse scalp neurofibroma had mildly enlarged since the scan performed 10 years prior (now 1.2 cm in depth, previously 1 cm).
Treatment¶
- Observation:
- Appropriate for asymptomatic, small neurofibromas
- Regular follow-up to monitor for growth or malignant transformation
- Surgical excision:
- Indicated for symptomatic lesions or those with suspected malignant transformation
- Complete resection with preservation of nerve function when possible
- Radiation therapy:
- Limited role due to potential for malignant transformation
- May be considered for inoperable tumours causing significant symptoms
- Targeted therapies:
- MEK inhibitors (e.g., selumetinib) show promise in reducing tumour size and improving symptoms in NF1-associated plexiform neurofibromas
- Regular surveillance:
- Annual clinical examinations and imaging studies for patients with NF1
- Monitor for development of new lesions and potential malignant transformation
Differential diagnosis¶
| Differential Diagnosis | Distinguishing Feature |
|---|---|
| Schwannoma | Typically encapsulated; often associated with larger nerves |
| Lipoma | Homogeneous fat signal on MRI; no enhancement |
| Ganglion cyst | Fluid-filled; no solid component on imaging |
| Lymph node | Hilar structure; different shape and location |
| Dermatofibroma | Typically smaller; confined to dermis |
| Leiomyoma | Originates from smooth muscle; different histology |
| Epidermal inclusion cyst | Contains keratin debris; no nerve involvement |
| Malignant peripheral nerve sheath tumour | Larger size; irregular borders; rapid growth |
| Plexiform neurofibroma | Involves multiple nerve fascicles; "bag of worms" appearance |
| Fibroma | No nerve involvement; different histology |