Spinoerebellar Ataxia (SCA)¶
Summary
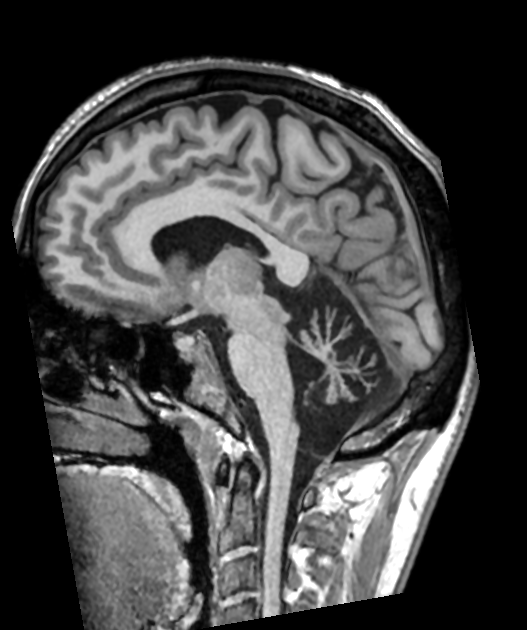
- Progressive neurodegenerative disorder affecting the cerebellum and spinal cord
- Characterised by ataxia, dysarthria, and oculomotor abnormalities
- Imaging reveals cerebellar atrophy, with variable involvement of brainstem and spinal cord
Pathophysiology¶
- Autosomal dominant inheritance in most cases
- Caused by expansion of CAG trinucleotide repeats in various genes
- Results in progressive loss of Purkinje cells and other neurons in the cerebellum and brainstem
- Different SCA subtypes (SCA1-SCA48) with distinct genetic mutations and clinical features
Demographics¶
- Prevalence: 1-5 per 100,000 worldwide
- Onset typically in adulthood, but can occur in childhood or late adulthood
- SCA3 (Machado-Joseph disease) is the most common subtype globally
- Geographical variations in subtype prevalence exist
Diagnosis¶
- Clinical presentation:
- Progressive ataxia
- Dysarthria
- Oculomotor abnormalities (nystagmus, slow saccades)
- Additional features vary by subtype (e.g., cognitive impairment, peripheral neuropathy)
- Genetic testing to identify specific SCA subtype
- Family history assessment
- Neurological examination
- Imaging studies to support diagnosis and exclude other causes
Imaging¶
- MRI is the modality of choice
- Key findings:
- Cerebellar atrophy, particularly of the vermis
- Pontine atrophy in some subtypes (e.g., SCA1, SCA2)
- Spinal cord atrophy, especially in SCA1 and SCA3
- T2/FLAIR hyperintensities in cerebellar white matter and middle cerebellar peduncles
- Advanced techniques:
- Diffusion tensor imaging (DTI) may show reduced fractional anisotropy in cerebellar white matter
- Voxel-based morphometry can quantify regional brain volume loss
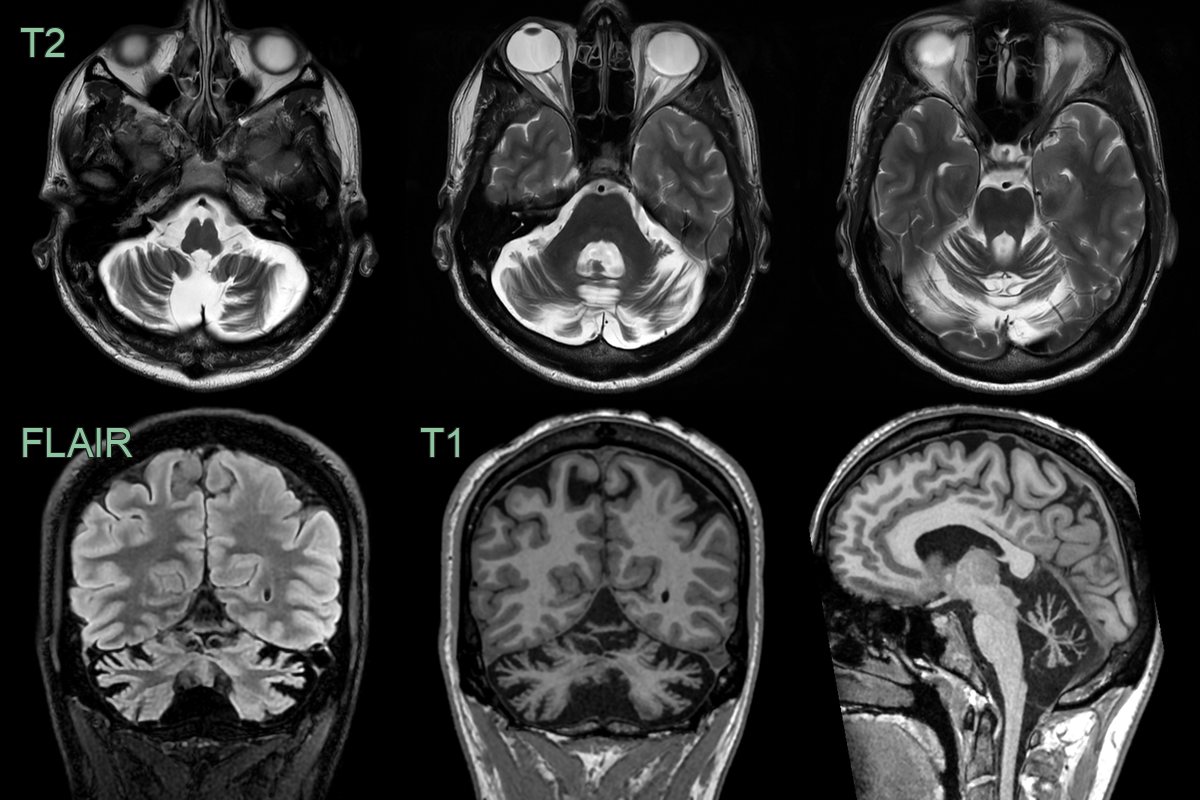
- A 35-year-old patient presented with progressive ataxia.
- MRI showed severe symmetrical atrophy of the cerebellar hemispheres and vermis.
- The ataxia genetic panel was negative (whole genome sequencing results are awaited).
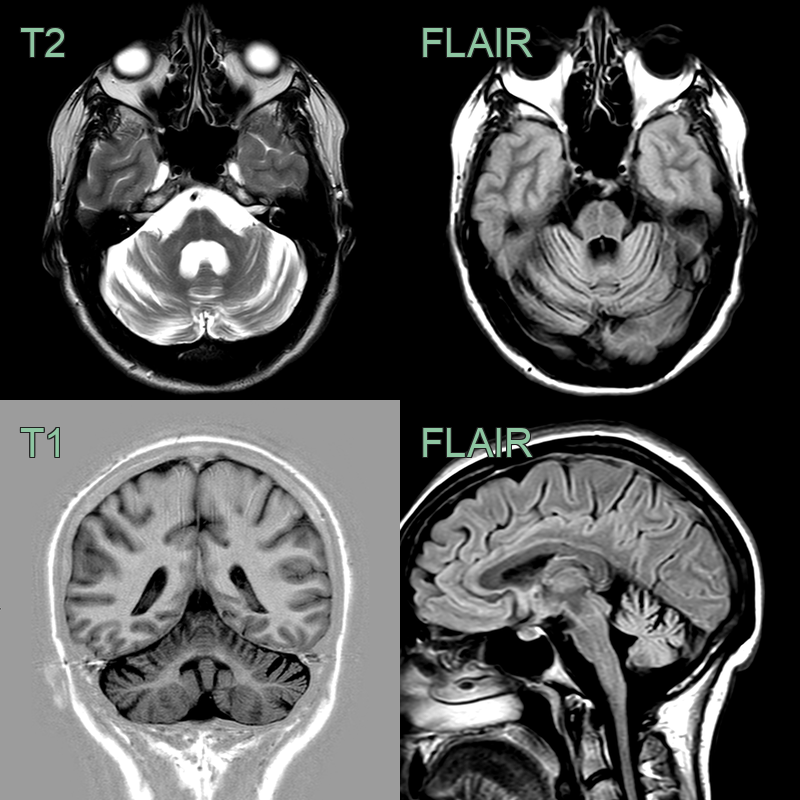
- A 40-year-old patient presented with slowly progressive ataxia.
- MRI showed severe cerebellar hemisphere, vermis, pontine atrophy.
- Atrophy of the cervical cord was less pronounced.
Treatment¶
- No curative treatment available
- Management focuses on symptomatic relief and supportive care:
- Physical therapy to improve balance and coordination
- Occupational therapy for activities of daily living
- Speech therapy for dysarthria
- Medications for specific symptoms (e.g., baclofen for spasticity)
- Genetic counselling for affected individuals and family members
- Emerging therapies:
- Antisense oligonucleotides to reduce mutant protein expression
- Stem cell therapy (experimental)
- Gene therapy approaches under investigation
Differential diagnosis¶
| Differential Diagnosis | Distinguishing Feature |
|---|---|
| Multiple System Atrophy (MSA-C) | "Hot cross bun" sign in pons on T2; putaminal rim sign; more diffuse olivopontocerebellar atrophy including inferior olives |
| Friedreich's Ataxia | Spinal cord atrophy with dorsal column T2 signal; relatively preserved pons; no middle cerebellar peduncle involvement |
| Paraneoplastic cerebellar degeneration | Cerebellar cortical atrophy with FDG-PET hypometabolism; may show early FLAIR signal in cerebellum before structural atrophy |
| Fragile X-associated Tremor/Ataxia Syndrome (FXTAS) | Symmetric T2 hyperintensity in middle cerebellar peduncles; cerebellar and cerebral white matter changes |
| CANVAS Syndrome | Cerebellar cortical atrophy with preserved brainstem volume; dorsal column spinal cord involvement |