Wilson's Disease¶
Summary
- Autosomal recessive disorder of copper metabolism
- Characterised by excessive copper accumulation in various organs, primarily liver and brain
- Imaging findings include basal ganglia abnormalities and hepatic cirrhosis
Pathophysiology¶
- Caused by mutations in ATP7B gene on chromosome 13
- Impaired biliary copper excretion
- Reduced incorporation of copper into ceruloplasmin
- Results in toxic accumulation of copper in liver, brain, cornea, and other organs
Demographics¶
- Worldwide prevalence: 1 in 30,000 to 1 in 100,000
- No gender predilection
- Higher prevalence in certain populations (e.g., Sardinia, Eastern Asia)
Diagnosis¶
- Clinical presentation:
- Hepatic dysfunction
- Neurological symptoms (e.g., tremor, dysarthria, dystonia)
- Psychiatric disturbances
- Kayser-Fleischer rings in cornea
- Laboratory findings:
- Low serum ceruloplasmin
- Elevated 24-hour urinary copper excretion
- Elevated hepatic copper concentration on liver biopsy
- Genetic testing for ATP7B mutations
Imaging¶
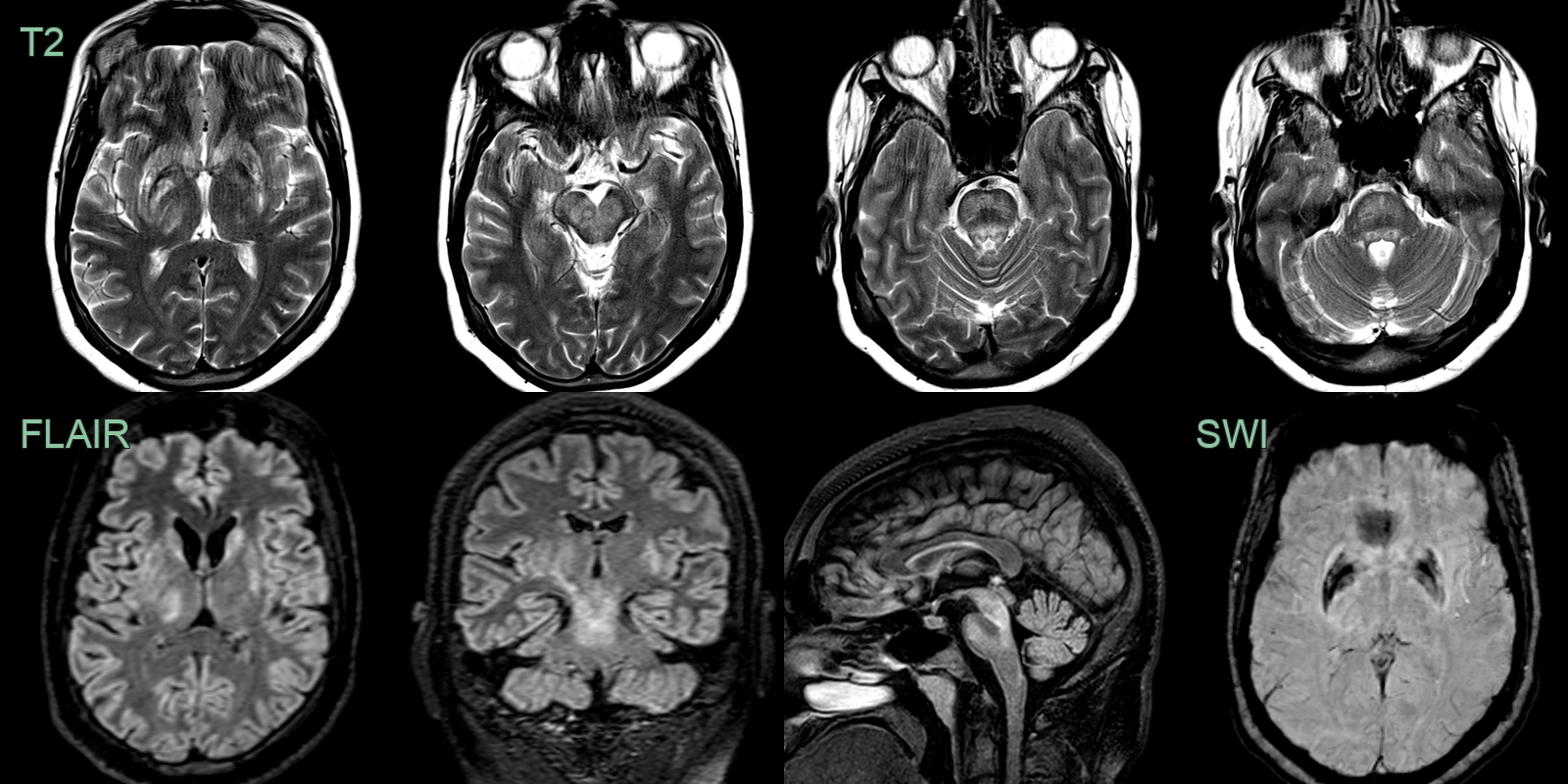
- A 25-year-old male presented 6 months prior with dysarthria, dystonia and cirrhosis.
- Imaging showed patchy hyperintensity in the deep grey nuclei and brainstem.
- SWI showed hypointensity in the globi pallidi.
Treatment¶
- Copper chelation therapy:
- D-penicillamine
- Trientine
- Zinc supplementation to reduce copper absorption
- Dietary copper restriction
- Liver transplantation for severe hepatic dysfunction or fulminant liver failure
- Symptomatic management of neurological and psychiatric manifestations
- Family screening and genetic counselling
Differential diagnosis¶
| Differential Diagnosis | Differentiating Feature |
|---|---|
| Acquired hepatocerebral degeneration | Bilateral symmetric T1 hyperintensity of globi pallidi; T2 hyperintensity in basal ganglia and white matter; no SWI hypointensity in copper-specific distribution |
| Leigh disease | Symmetric T2 hyperintensity in basal ganglia and brainstem periaqueductal grey; putamen and caudate involvement; no SWI hypointensity |
| Japanese encephalitis / flaviviral encephalitis | Bilateral thalamic and basal ganglia T2 hyperintensity; may show haemorrhage and restricted diffusion |